
一、工艺规程
工艺规程 为生产特定数量的成品而制定的一个或一套文件,包括生产处方、生产操作要求和包装操作要求,规定原辅料和包装材料的数量、工艺参数和条件、加工说明包括中间控制、注意事项等内容。
(一)原料药生产工艺规程
1.品名、产品概述、化学反应及副反应过程,生产工艺及设备流程图。
2.工艺流程及生产操作要求。包括:①物料、中间产品名称及代码,投料量、投料比;②工艺过程及参数,操作顺序及要求;③物料、中间产品储存条件及期限,标签、包装材料。
3.生产过程的质量控制,物料、中间产品、成品的质量标准,取样方法。
4.生产地点、设备含仪表包括型号及材质一览表主要设备生产能力。
5.预防措施及注意事项,技术安全及防火、劳动保护,原料消耗定额和技术经济指标,副产品、回收品的处理,“三废”治理及排放标准。
6.操作工时与生产周期,单个步骤或整个工艺过程的时限,劳动组织与岗位定员。
7.附录包括有关理化常数、曲线、图表、计算公式、换算表等。
(二)制剂生产工艺规程
1.生产处方 包括:①产品名称和产品代码;②产品剂型、规格和批量;③所用原辅料清单,包括生产过程中使用但不在成品中出现的物料,阐明每一物料的指定名称、代码和用量,如原辅料的用量需要折算时,还应当说明计算方法。
2.生产操作要求 包括:①对生产场所和所用设备的说明如操作间的位置和编号、洁净度级别、必要的温湿度要求、设备型号和编号等;②关键设备的准备如清洗、组装、校准、灭菌等所采用的方法或相应操作规程编号;③详细的生产步骤和工艺参数说明如物料的核对、预处理、加入物料的顺序、混合时间、温度等;④所有中间控制方法及标准;⑤预期的最终产量限度,必要时,还应当说明中间产品的产量限度,以及物料平衡的计算方法和限度;⑥待包装产品的储存要求,包括容器、标签及特殊储存条件;⑦需要说明的注意事项。
3.包装操作要求 包括:①以最终包装容器中产品的数量、重量或体积表示的包装形式;②所需全部包装材料的完整清单,包括包装材料的名称、数量、规格、类型以及与质量标准有关的每一包装材料的代码;③印刷包装材料的实样或复制品,并标明产品批号、有效期打印位置;④需要说明的注意事项,包括对生产区和设备进行的检查,在包装操作开始前,确认包装生产线的清场已经完成等;⑤包装操作步骤的说明,包括重要的辅助性操作和所用设备的注意事项、包装材料使用前的核对;⑥中间控制的详细操作,包括取样方法及标准;⑦待包装产品、印刷包装材料的物料平衡计算方法和限度。
二、操作规程
操作规程 是指经批准用来指导设备操作、维护与清洁、验证、环境控制、取样和检验等药品生产活动的通用性文件,也称标准操作规程,英文缩写为“SOP”。
操作规程内容包括:①操作名称;②编写依据;③操作范围及条件,应注明时间、地点、对象、目的;④操作步骤或程序包括准备过程,操作过程、结束过程;⑤操作标准;⑥操作结果的验收、检验标准;⑦操作过程复核与控制;⑧操作过程的安全事项与注意事项;⑨操作中使用的物品、设备、器具及其编号;⑩操作异常情况处理等。
范例:某车间B级区容器、器具清洁消毒标准操作规程
| 题目 | 车间B级区容器、器具清洁消毒标准操作规程 | |||
| 颁发部门 | 生产部 | 分发部门 | 质保部、某车间 | |
| 制定: | 年 月 日 | 审核: | 年 月 日 | 编号:SOP-SM011-01 |
| 批准: | 年 月 日 | 生效日期 | 年 月 日 | 共1页第l页 |
| 目 的:建立某车间B级区容器、器具清洁消毒标准操作规程,保证工艺卫生,防止污染。 应用范围:某车间B级区(含局部A级)容器、器具的清洁消毒操作。 责任人:操作工、QA员。 内 容 1.清洁工具 毛刷、试管刷、丝光毛巾。 2.清洁地点 容器具清洗间。 3.清洁剂及用量 0.1%洗洁精溶液、1%氢氧化钠溶液,擦拭用量每平方米不得低于25毫升。 4.消毒剂及用量 纯蒸汽、75%乙醇,擦拭用量每平方米不得低于25毫升。 5.清洁消毒频次及方法 5.1玻璃容器(包括试剂瓶、量筒、锥形瓶等) 5·1·1每次使用后用毛刷蘸取少许0.1%洗洁精溶液,将内外壁刷洗一遍,然后边用水冲边刷洗,直到洗净为止。最后用少量注射用水刷洗3次。 5·1·2测定装量的量简每天生产结束后,按5.1.1进行清洁后,在3%双氧水溶液中浸泡15分钟,最后用注射用水刷.洗3次,擦干,备用。 5.2塑料容器具、胶塞周转桶、回收料桶等不可高温灭菌制品 5.2.1每次使用后立即用经0.22微米过滤的注射用水洗刷干净后控干,备用。 5·2·2每天生产结束后及第一次使用前用过滤注射用水洗刷干净,控干后,用75%乙醇全面擦拭,待消毒剂滞留l5分钟后,用过滤注射用水反复刷洗去除消毒剂残留,倒置控干,备用。 5.3触药小不锈钢工具及容器(包括灌注系统中不锈钢制品)。 每天生产结束后,用过滤的注射用水清洁干净后,放入不锈钢容器中进行湿热灭菌,121。Cl5分钟,备用。 5.4工器具(包括不锈钢扳手等) 每次使用后立即用过滤注射用水洗刷干净后控干,备用。污迹不易去除时,可用1%氢氧化钠溶液去除污迹后,再用注射用水反复冲洗去除清洁剂残留后控干,备用。使用前用75%乙醇溶液擦拭进行消毒。自然晾干后使用。 6.清洁效果评价 目测检查应无可见污迹和残留物。 7.清洁工具的清洁及存放 使用后的清洁工具按“一车间洁净区清洁工具清洁消毒标准操作规程’,进行清洁操作。 8.容器、器具的存放 凡已清洁的容器、器具,不宜套装放置,倒置放于容器存放问,并贴挂“已清洁’’状态标识。 9.容器、器具清洁后有效期 9.1连续生产时,有效期为48小时。 9·2若停用超过48小时,下次使用前按5.1、5.2和5.3、5.4对其进行清洁消毒操作。 10.生产结束至开始进行清洁操作等待时间不得超过2小时。 | ||||
三、批记录
批记录 用于记述每批药品生产、质量检验和放行审核的所有文件和记录,可追溯所有与成品质量有关的历史信息。
批记录包括:批生产指令记录、批包装指令记录、批生产记录和批包装记录、批检验记录、批放行记录。
生产中批指令一般包括批生产指令和批包装指令。
(一)批生产指令
生产指令包括以下主要内容:①产品名称及代码、规格、批号、生产日期;②计划产量;③生产技术依据、生产操作规程编号;④专用模具代码清单;⑤原辅料定额量、计划领用量;⑥生产技术部门负责人、生产车间生产负责人签名批准。
(二)批生产记录
批生产记录包括以下主要内容:①产品名称、代码、规格、批号;②生产以及中间工序开始、结束的日期和时间;③每一原辅料的批号、投料量、折算投料量、实际投料量包括投入的回收或返工处理产品的批号及数量、称量人与复核人签名;④所用主要生产设备的编号;⑤相关生产操作或活动、工艺参数及控制范围,各步生产过程控制操作记录,操作者签名及签日期时间;⑥中间控制结果的记录以及操作人员的签名;⑦各生产阶段及不同工序的产品数量,物料平衡的计算;⑧结退料记录;⑨设备清洁、操作、保养记录;⑩前次包装操作的清场记录副本及本次包装清场记录正本;⑪对特殊问题或异常事件的记录,包括对偏离工艺规程的偏差情况的详细说明或调查报告处理及结果记录,并经签字批准。
范例:大容量注射剂称量、浓配批生产记录
| 产品名称 | 产品规格 | 产品批号 | |||||||
| 配制量 | 万毫升 | 理论产量 | 瓶 | 生产日期 | 年 月 日 | ||||
| 生产依据 | 本品种工艺规程 | ||||||||
操作 步骤 | 操作指令 | 操作记录 | |||||||
| 生 产 前 检 查 | 1.生产文件、清场合格证 2.生产现场 3.设备、容器具、管道 4.工艺用水、电、气 5.仪器、仪表 6.检查完毕,符合规定,更换状态标识 7.洁净区温度和相对湿度 | 口齐全 口已清洁 口完好口已清洁 口供应充足 口已校正,在有效期内 口已清洁 口已更换 口符合规定 温度: ℃相对湿度 % | |||||||
| 检查人 | 检查时间 | ||||||||
|
准备 | 8.根据批生产指令领取所需原辅料,核对品名、规格、编号、批号、数量等 9.冲洗浓配罐、管路及容器具 10·配制pH调节剂(10%盐酸或l0%氢氧化钠):( )毫升 | 口已核对,符合要求 领料人: 复核人: 清洁情况:口合格 配制情况:口合格 操作人: 复核人: | |||||||
| 外清 | 11·将领取的原辅料外包装在外清间逐件擦净,移入缓冲间 | 检查情况:口合格 操作人: 质检员: | |||||||
| 称量 | 12·按一车间称量、浓配操作程序称量项下规定,逐一称量核对原辅料 | 称量情况:口合格 称量过程记录详见范例 操作人: 复核人: | |||||||
浓配 | 13.按本品种配制SOP进行操作 14·加入规定量的注射用水,加热升温投入指令量原料 15.加入pH调节剂和指令量活性炭 16.加热煮沸15分钟 | 口已操作,符合SOP 加注射用水: 升投料时间: 加炭时间: 加pH调节剂量: 毫升 煮沸时间: 至 续煮时间: 分钟 操作人: 复核人: | |||||||
脱炭 循环 | 17.脱炭自身循环至取样目视药液澄清,过滤至稀配罐内 | 脱炭循环时间: 分钟 过滤打料时间: 至 稀配罐号: 操作人: 复核人: | |||||||
物料 结存 | 18.将剩余的物料退库或下批使用 | 口退库 口下批使用 操作人: 复核人: | |||||||
| 清场 | 19.见清场记录 | 口批清场 口日清场 | |||||||
| 备注 | |||||||||
(三)批包装指令
批包装指令包括以下主要内容:①包装产品名称及代码、包装规格、批号、生产日期;②计划产量;③包装方法、包装要求、作业顺序、SOP编号、生产地点、使用设备与生产线及其编号;④专用模具代码清单;⑤包装材料定额量、计划领用量;⑥生产技术部门负责人、生产车间生产负责人签名批准。
(四)批包装记录
批号,发放和实际使用的数量;⑦根据工艺规程所进行的检查记录,包括中间控制结果;⑧包装操作的详细情况,包括所用设备及包装生产线的编号;⑨所用印刷包装材料的实样,并印有批号、有效期及其他打印内容,不易随批包装记录归档的印刷包装材料可采用印有上述内容的复制品;⑩包装操作记录,设备、生产线操作记录,操作者、复核者、负责人签名;⑪各生产阶段及不同工序的产品数量,物料平衡的计算;⑫结退料记录;⑬设备清洁、操作、保养记录;⑭前次包装操作的清场记录副本及本次包装清场记录正本;⑮特殊问题或异常事件的记录,包括对偏离工艺规程的偏差情况的详细说明或调查报告,并经签字批准。
(五)批检验记录
所有原料、辅料、包装材料、中间产品和成品都必须经过检验,确保符合相应标准,并有记录,检验记录包括:①原辅料、与药品直接接触的包装材料的检验项目,检验结果报告单;②中间产品检验项目,检验结果报告单;③成品检验项目,检验结果报告单。
(六)放行审核记录
放行审核记录包括对物料和产品的放行,产品包括药品的中间产品、待包装产品和成品。
物料与产品的放行应符合放行要求,详见模块三项目十任务四项下内容。
中间产品、待包装产品和成品放行应进行合规性审查,对药品生产现场的人员操作、清洁及环境监测过程进行核查,审核批生产、批包装、批检验等记录,实施电子记录的,应对电子记录予以审核和确认。对于批记录审核合格的,须经受权人或质量管理负责人批准放行,中间产品可进入下工序或使用,成品可发运投放市场。
四、电子记录
(一)术语
电子生产记录(以下简称EPR) 实施计算机化系统管理的企业在执行控制配方和生产操作过程中存储来自生产相关活动中的数据和信息,数据和信息可由系统产生以及人工输入或单纯人工输入。电子数据可能保存在一个或多个系统或数据库中。一般有两种类型:电子批生产记录(以下简称EBR)和电子设备历史记录(以下简称EDHR)。
EBR 用来记录一个批次,含回收、重新加工、返工批次,或连续过程中药品生产和质量控制的电子数据信息。
EDHR 一系列的记录涵盖一个已完成的电子设备的历史的记录。
(二)电子记录审核
电子记录审核指对从生产相关的运行中得到的数据进行筛选,用于编制审核与事务处理例如发布、隔离、拒绝报告的方法,该方法既包括对关键工艺异常的审核,也包括减少或取消对可接受数据和趋势的审核。
EPR审核适用于生产的全过程或工艺步骤的一部分,计算机化系统或与纸质记录形式共同使用的情形。
审核可采用人工审核和系统附带的自动审核功能。
在完整的生产报告使用数据审核之前,可以用分阶段的方法来实施数据审核。如果有程序化的技术控制措施来控制管理方法,可将每个报告作为整体报告一部分进行管理,而各个单独的报告和数据审核报告可以结合使用。
电子记录审核确保达到以下要求:将工艺保持在规定的可按受偏差范围内;准确地记录了数据与事件;当超出了规定的偏差范围或其他操作规定时,发出警报和得出警告;电子记录是准确、可信且安全的,用来进行流程产品审核的生产报告是准确的。
(三)电子记录数据库
生产过程中包含了大量主要数据,比如物料规格说明、工艺参数、警报与警告限制,或者由几个系统共同控制的工艺步骤程序。
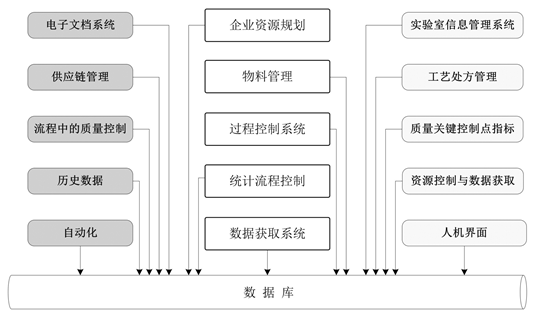
(四)电子表格
可以用应用程序工具来创建各种最终用户应用程序,包括定制的统计数据分析、本地数据库创建、数据挖掘、多元分析。
电子表格的应用类型包括:一次性电子表格、存档电子表格、数据库电子表格、模板电子记录表格、桌面数据库。
EPR审核、迁移、实时发布流程见图8–4。
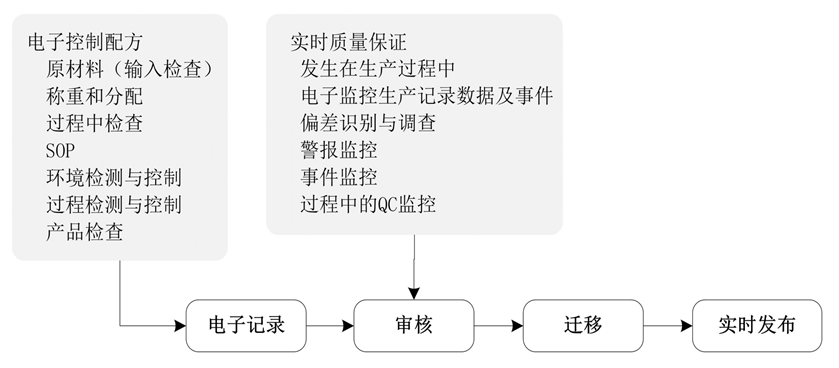
图8–4 电子记录审核、迁移、实时发布流程图

