
第一节 分 子 病
分子病(molecular disease)是指由于基因突变导致蛋白质的分子结构或合成数量异常,进而引起机体功能障碍的一类疾病。分子病这一名词是1949年美国化学家Pauling在研究镰形细胞贫血症时提出的,他发现患者异常血红蛋白β链N端的第6位谷氨酸被缬氨酸所替代,并把它称为血红蛋白S。迄今已发现的异常血红蛋白有700多种。分子病除了血红蛋白病以外,还有各种血浆白蛋白病、免疫球蛋白缺陷性疾病、凝血因子病、受体蛋白病等。
一、正常血红蛋白及其遗传控制
(一)正常血红蛋白的组成、种类和发育演变
血红蛋白是高等生物体内负责运载氧并维持血液酸碱平衡的一种蛋白质。正常血红蛋白(hemoglobin,Hb)是由珠蛋白和血红素组成的复合蛋白质。人体内血红蛋白为四聚体分子,每个单体分子是由一条珠蛋白肽链和一个血红素辅基组成的。肽链在生理条件下会盘绕折叠成球形,把血红素辅基包在里面,这条肽链盘绕成的球形结构又被称为珠蛋白。一个四聚体包括两条类α链(α链和ζ链)和两条类β链(ε链、γ链、δ链和β链),α链由141个氨基酸组成,β链由146个氨基酸组成。这6种不同的珠蛋白链可组合成人类的6种不同的血红蛋白,即Hb GowerⅠ(ζ2ε2)、Hb GowerⅡ(α2ε2)、Hb Portland(ζ2γ2)、Hb F(α2γ2)、Hb A(α2β2)和Hb A2(α2δ2)(表8-1)。
表8-1 不同发育阶段人体血红蛋白组成
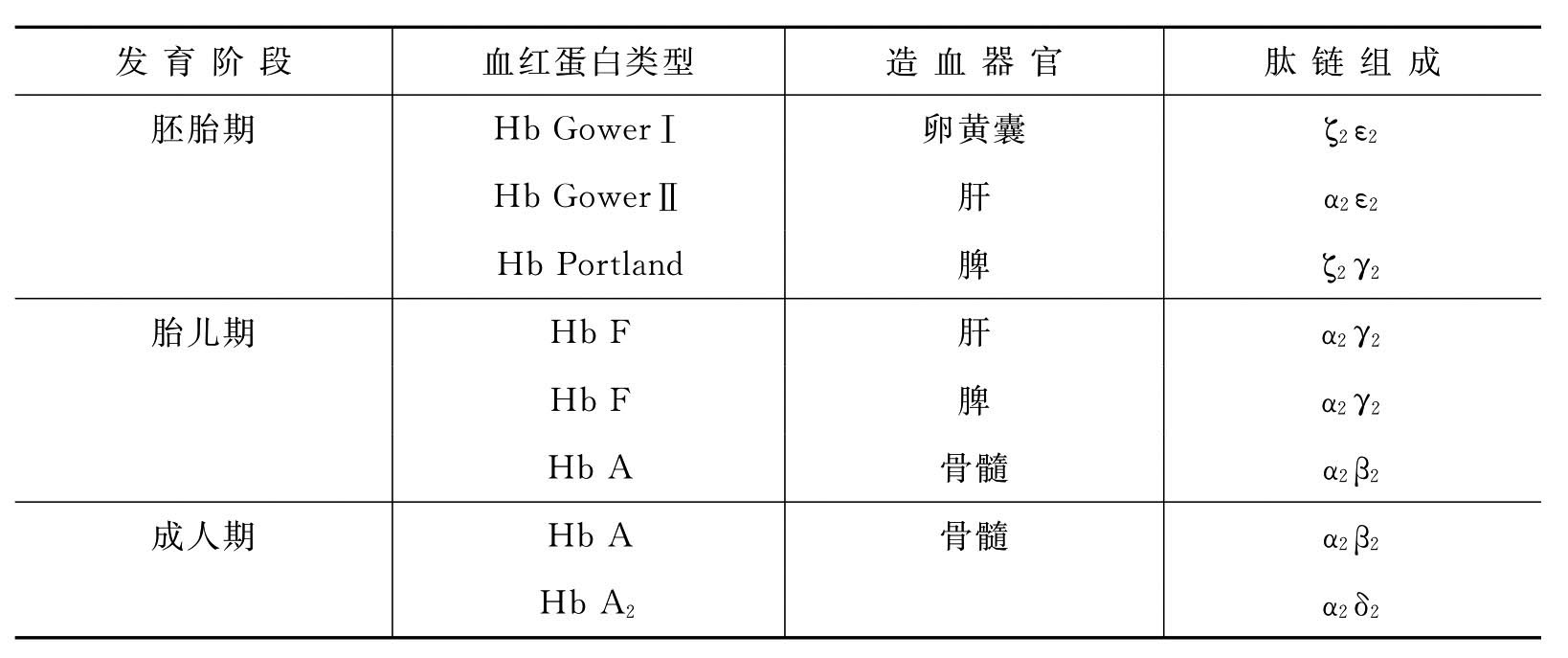
不同血红蛋白的携氧、释氧能力是不同的,在人体发育的不同阶段,会由不同的组织器官合成不同的血红蛋白满足机体发育的需求,血红蛋白的出现规律见图8-1。在胚胎初期合成的血红蛋白是Hb GowerⅠ、Hb GowerⅡ和Hb Portland。胎儿期(从妊娠第8周到出生)的血红蛋白主要是Hb F。出生后,随着β链合成迅速增加,γ链合成减少,所以成人有三种血红蛋白:Hb A约占97.5%;Hb A2约占2%;Hb F约占0.5%。胚胎期造血部位主要在卵黄囊,胎儿期主要在肝、脾,成人期则主要在骨髓。
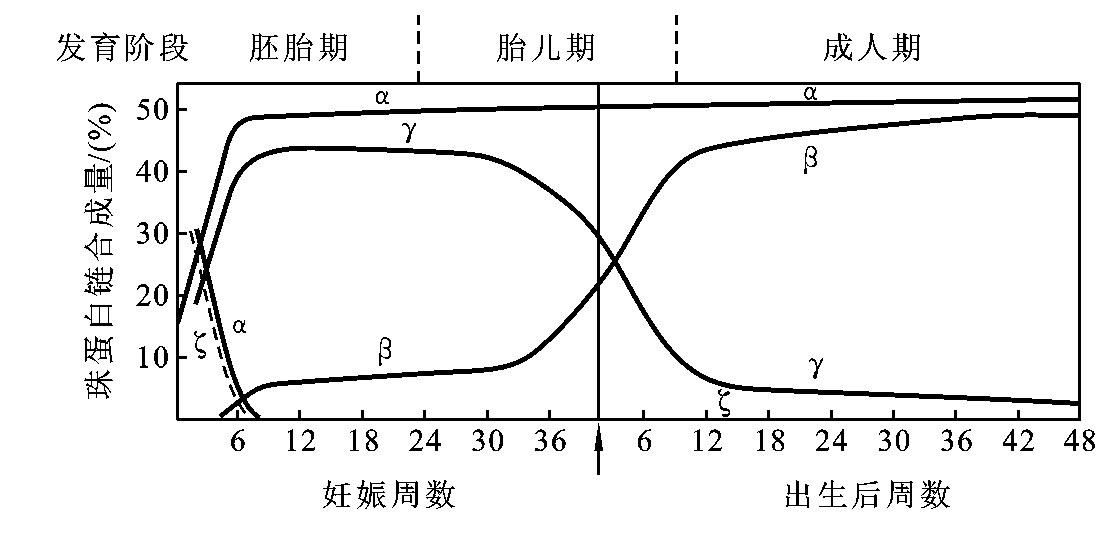
图8-1 血红蛋白的出现规律
(二)珠蛋白基因结构及其表达
1.珠蛋白基因结构 人类珠蛋白基因由α珠蛋白基因簇和β珠蛋白基因簇组成,α珠蛋白基因簇位于第16号染色体短臂上,包括7个基因,分别为ζ、Ψζ1、Ψζ2、Ψα2、Ψα1、α2、α1和θ。β珠蛋白基因簇位于第11号染色体短臂上,包括有6个基因,分别是ε、Gγ、Aγ、Ψβ、δ和β。
α珠蛋白基因簇和β珠蛋白基因簇的各基因结构很相似,都含有3个外显子和2个内含子(IVS1和IVS2)。α珠蛋白基因簇中的IVS1位于31和32密码子之间,IVS2位于99和100密码子之间(图8-2)。β珠蛋白基因簇中的IVS1位于30和31密码子之间,IVS2位于104和105密码子之间。人类β珠蛋白基因簇的结构见图8-3。
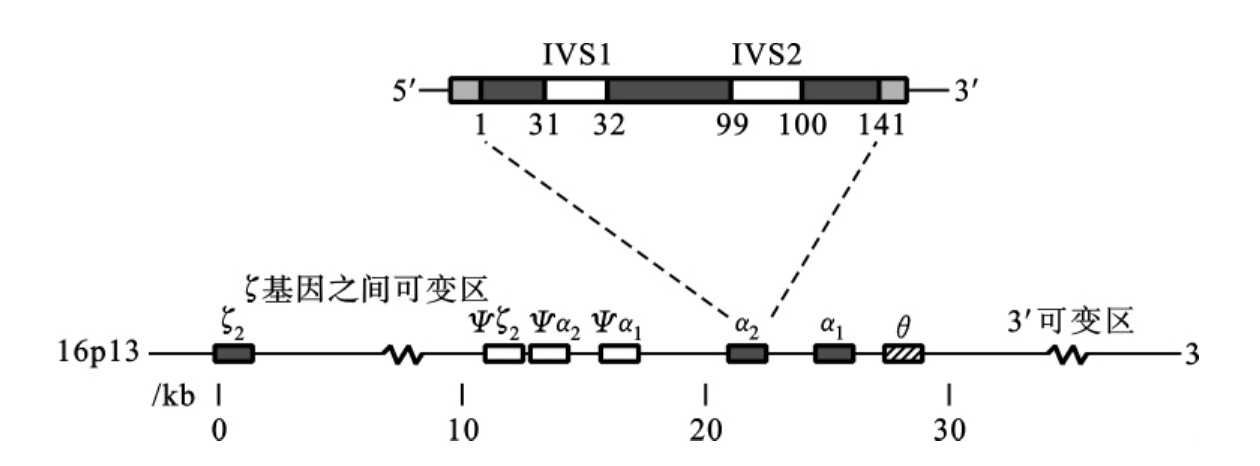
图8-2 人类α珠蛋白基因簇的结构示意图
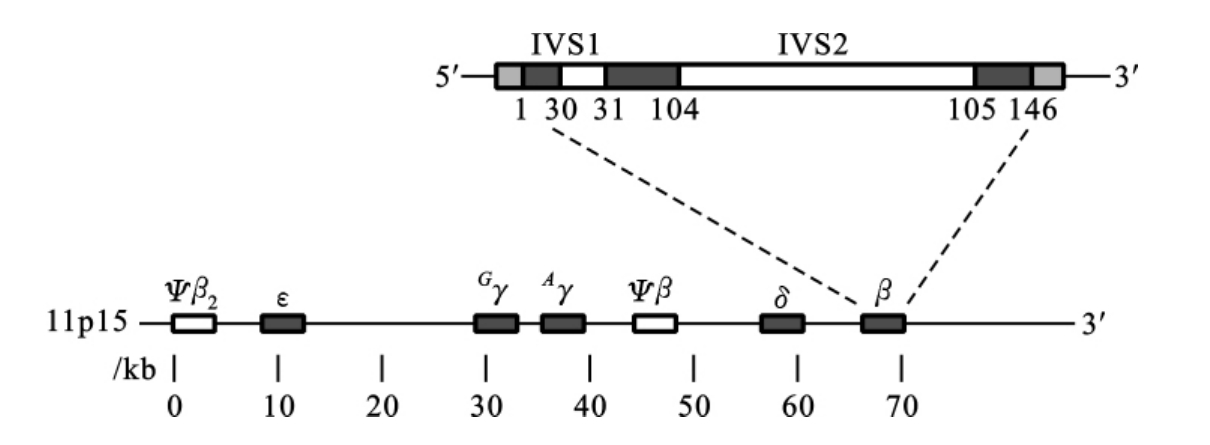
图8-3 人类β珠蛋白基因簇的结构示意图
2.珠蛋白基因的表达 珠蛋白基因的表达过程与其他真核生物的基本相似。首先细胞核内的珠蛋白基因先转录出一个分子量相对较大的mRNA前体,再经过戴帽、添尾等加工过程,然后在相关酶的作用下切去内含子,将外显子连接起来,形成成熟的mRNA。成熟的mRNA从细胞核进入细胞质,并与核糖体结合,经过翻译,形成相应的珠蛋白肽链,最后结合成各种血红蛋白。
二、血红蛋白病
血红蛋白病(hemoglobinopathy)是指珠蛋白基因突变导致珠蛋白合成异常所引起的疾病,通常分为两大类,即异常血红蛋白病和地中海贫血。
(一)异常血红蛋白病
异常血红蛋白病是指由于珠蛋白基因突变导致组成珠蛋白的肽链在结构上和功能上发生异常,伴有临床表现的,称为异常血红蛋白病。异常血红蛋白种类较多,全世界已报道的有750种以上。我国发现的有70余种,其中31种是世界首次报道。
1.异常血红蛋白病的基本类型
(1)镰形细胞贫血症:发病原因是患者血红蛋白β链第6位的谷氨酸被缬氨酸取代,使血红蛋白A变成血红蛋白S,导致血红蛋白分子表面电荷改变,溶解度降低。血红蛋白S在脱氧状态下聚集成长棒状,使红细胞镰变。本病呈常染色体隐性遗传,在非洲和北美洲黑种人群中患病率较高。杂合子一般无临床症状,但在氧分压低时可引起红细胞镰变,称为镰形细胞性状。纯合子症状严重,镰变细胞使血液黏性增加,微血管易栓塞,造成散发性局部组织缺氧,产生疼痛,甚至坏死,如腹部疼痛、心肌梗死等,还会引发严重溶血性贫血及脾肿大等症状。
(2)血红蛋白M病:本病又称血红蛋白M遗传性高铁血红蛋白血症。正常血红蛋白血红素中的铁原子通过与珠蛋白链上特定的氨基酸连接和作用,保证二价铁离子(Fe2+)的稳定,维持与氧的亲和力。血红蛋白M由于发生碱基置换,使珠蛋白链中与铁原子连接的有关氨基酸发生了替代,导致部分血红素的Fe2+变成Fe3+,呈高铁状态,影响了正常的携氧功能,造成组织缺氧。患者出现发绀症状,并导致继发性红细胞增多。
(3)不稳定血红蛋白病:本病是指由于珠蛋白基因突变使珠蛋白链上的氨基酸排列顺序发生改变,导致分子结构不稳定的血红蛋白病。不稳定血红蛋白容易自发或在氧化剂作用下降解为单体,易与血红素分离。失去血红素的珠蛋白链可沉淀,形成不溶性珠蛋白小体,并附着于红细胞膜,使细胞膜可塑性降低,易发生血管内、外溶血。已知的不稳定血红蛋白病有130多种,多为常染色体显性(或不完全显性)遗传,患者多为杂合子。主要表现为溶血性贫血,轻重程度不一,重者可发生溶血危象而危及生命。如Hb Bristol不稳定血红蛋白病是指由于β链第67位缬氨酸被天冬氨酸取代,使血红蛋白分子结构不稳定的一种血红蛋白病,临床症状有先天性溶血性贫血、黄疸和脾肿大。
(4)氧亲和力改变的异常血红蛋白病:本病是由于多肽链上氨基酸的替换而使血红蛋白分子与氧的亲和力升高或降低,导致携氧功能改变。例如Hb Rainer(β145酪→半胱),氨基酸替换后,血红蛋白分子与氧的亲和力增高,运送给组织的氧减少,使红细胞增多;又如Kansas(β102冬胺→苏),替换后的血红蛋白分子与氧的亲和力降低,使动脉血的血氧饱和度下降,重者可引起发绀症状。
2.异常血红蛋白病的分子基础 异常血红蛋白的形成是珠蛋白基因突变的结果,涉及多种突变类型,主要类型如下。
(1)单个碱基置换:单个碱基置换是突变中最普遍的一种。90%以上的异常血红蛋白病都是由于珠蛋白基因发生单个碱基置换所引起的。其中错义突变较常见,例如,镰形细胞贫血症是由于β珠蛋白基因的第6位密码子由GAA变成GUA,使谷氨酸变为缬氨酸。此外,若终止密码子发生单个碱基置换,会使肽链延长,若编码一个氨基酸的密码子变成终止密码子,会使肽链缩短。以上这些都会形成异常血红蛋白,如Hb Constant Spring和Hb Mckees Rocks。
(2)移码突变:由于在合成血红蛋白的基因中插入或丢失一个或几个(不是3的倍数)碱基,将导致在插入或缺失点以后的密码子位移,使翻译出的氨基酸排列顺序也发生相应改变,如Hb Wayne是由于α链第138位密码子UCC丢失了一个C,使缺失点后的碱基顺序发生移位,重新编码,原来第142位上的终止密码子变成可读密码子,直到在第147位遇到下一个终止密码子翻译才停止。
(3)整码突变:整码突变是指在mRNA上插入或缺失一个或多个密码子,使编码的肽链比正常肽链缩短或延长。如Hb Grady是α链第116位的脯氨酸后边插入了苯丙氨酸、苏氨酸、脯氨酸三个氨基酸。
(4)不等交换:不等交换是指在减数分裂时编码两条不同肽链的基因所在的染色体发生了错位联会,进行互换,结果形成两种不同的融合基因(fusion gene),如:Hb Lepore的类β链的N端与δ链相同,C端与β链相同,故称为δβ链;Hb anti-Lepore的N端与β链相同,C端与δ链相同,故称为βδ链。
(二)地中海贫血
由于珠蛋白基因突变,使血红蛋白中的珠蛋白肽链有一种或几种合成减少或不能合成,导致血红蛋白的组成成分发生改变所引起的一种溶血性贫血,称为地中海贫血,也称珠蛋白生成障碍性贫血。地中海贫血主要有α地中海贫血、β地中海贫血,还有比较少见的δβ地中海贫血和γδβ地中海贫血。
1.α地中海贫血 α地中海贫血简称α地贫,是指α链合成减少或完全不能合成而引起的溶血性贫血。人类第16号染色体上各有2个α珠蛋白基因,一对染色体上共有4个α珠蛋白基因,大多数α地中海贫血是由于α珠蛋白基因缺失所致,少数为点突变造成的。若仅是一条染色体上的1个α基因缺失,则α链的合成部分受抑制,称为α+地中海贫血,若一条染色体上的2个α基因均缺失或缺陷,称为α0地中海贫血。
1)α地中海贫血的分子基础 引起α地中海贫血基因突变的方式主要有缺失突变和点突变,缺失突变较常见。
(1)缺失突变:位于第16号染色体上的α珠蛋白基因簇内有α1和α2两个基因,该基因簇内可发生长短不一的基因缺失,导致α珠蛋白肽链合成减少。
(2)点突变:α珠蛋白基因的点突变类型很多,部分点突变会导致α珠蛋白肽链合成减少,引发的α地中海贫血称为非缺失型α地中海贫血。
2)α地中海贫血的临床分类 根据临床表现的严重程度的不同,将α地中海贫血分为4种类型。
(1)Hb Bart’s胎儿水肿综合征:患者第16号染色体上4个α基因缺失,基因型表示为(--/--),完全不能合成α珠蛋白肽链,因而不能形成胎儿Hb F。相对过多的γ链聚合成Hb Bart’s(γ4)。Hb Bart’s对氧的亲和力很高,因而释放到组织的氧很少。因组织严重缺氧导致胎儿全身水肿,造成胎儿子宫内死亡或新生儿死产。胎儿双亲均为α0地中海贫血杂合子(--/αα),他们再生患儿的风险为1/4。因此,已生患儿的夫妇应做产前诊断。
(2)血红蛋白H病:患者为α0地中海贫血和α+地中海贫血的杂合子,基因型为(--/-α),有3个α基因缺失,或是(--/ααT),有2个α基因缺失,一个基因存在点突变(αT代表有突变)。缺失3个α基因,只能合成少量α链,相对过剩的β链自身聚合成四聚体Hb H(β4)。Hb H结构很不稳定,易被氧化而解聚成单链,并沉淀积聚成包涵体附着于红细胞膜上,使红细胞柔韧性降低,引起中度或重度溶血性贫血。患者双亲基因型多数为(--/αα)和(-α/αα),少数为(--/αα)和(αα/ααT),其子女发病风险为1/4。
(3)轻型α地中海贫血:本病又称标准型α地中海贫血,患者为α0地中海贫血的杂合子(--/αα)或α+地中海贫血纯合子(α-/α-),有2个α基因缺失。由于能合成一定量的α珠蛋白链,所以患者多无临床表现或有轻度溶血性贫血。两个轻型α地中海贫血患者婚配,生育Hb Bart’s水肿胎儿的概率为1/4。
(4)静止型α地中海贫血:患者为α+地中海贫血杂合子(α-/αα),仅有一个α基因缺失,这种个体的α珠蛋白肽链与β珠蛋白肽链合成数量相当,往往无临床症状。静止型α地中海贫血与轻型α地中海贫血患者婚配,有1/4的概率生育血红蛋白H病的患儿。
2.β地中海贫血 β地中海贫血简称β地贫,主要由于基因发生点突变或基因缺失形成。β地中海贫血的基因突变种类较多,迄今已发现的突变点已达100多种,国内已发现28种。中国人部分β地中海贫血的基因突变类型见表8-2。其中常见的基因突变类型有以下3种。①无功能mRNA突变:a.无义突变,如密码子CD 17(A→T),突变后形成了无功能的mRNA;b.移码突变,如密码子CD 41/42(-TCTT)和密码子71/72(+A),该突变形成的mRNA稳定性降低或失去功能;c.起始密码突变,如ATC→AGG,该突变形成异常的mRNA。②RNA加工障碍突变:a.剪接位点改变,如IVS-1(1位G→T),突变后失去一个剪接信号,形成长于正常值的mRNA;b.共有序列改变,如IVS-1(5位G→C),形成剪接异常的mRNA;c.IVS内部改变,如IVS-2(654位C→T),在突变部位形成一个新的剪接信号,产生异常的mRNA。③转录调控区突变,如密码子CD-28(A→G),该突变主要发生在启动子区,使基因转录水平下降,mRNA生成减少。基因缺失和有些点突变可使β链的合成完全受抑制,称为β0地中海贫血。有些突变使β链合成部分受抑制,则称为β+地中海贫血。
表8-2 中国人部分β地中海贫血的基因突变类型

续表
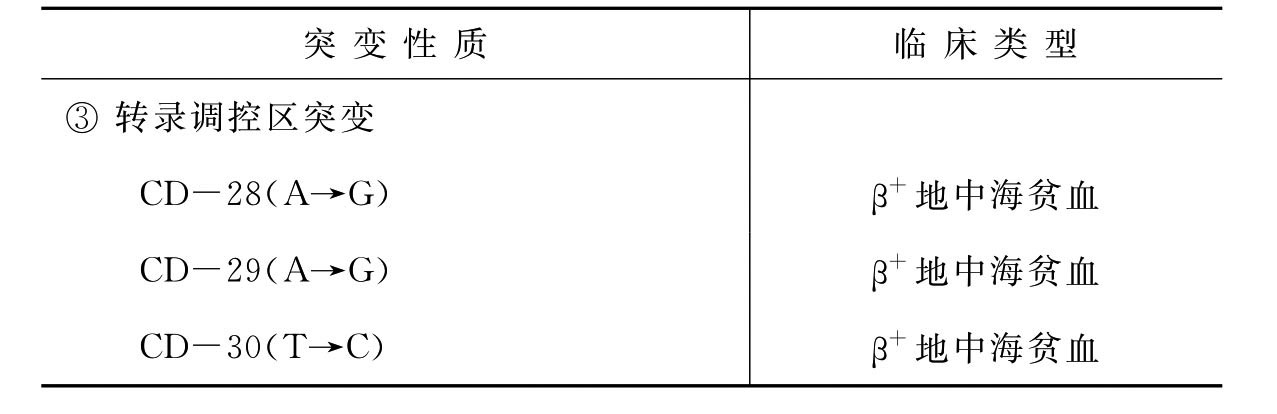
注:CD表密码子;IVS表内含子。
1)β地中海贫血的分子基础 根据点突变发生位置的不同,主要分为5种类型。
(1)编码区内的无义突变、移码突变和起始密码子突变:使合成的mRNA稳定性降低或功能丧失,不能合成正常的β珠蛋白肽链,多数可形成β0地中海贫血。
(2)内含子突变:突变如果发生在外显子和内含子交界处的保守序列,会影响内含子的正常剪接,产生异常的mRNA。
(3)外显子突变:某些外显子突变后,可在突变部位形成一个新的剪接点,产生剪接位点异常的mRNA。
(4)启动子突变:主要发生在β珠蛋白基因5′端的转录调控区,使基因转录效率降低、β珠蛋白mRNA合成减少,导致β+地中海贫血。
(5)加工修饰位点突变:戴帽和添尾对mRNA的形成是非常重要的。如果戴帽部位和多聚腺苷酸信号位点发生突变,那么转录产物不能准确裂解,产生不稳定mRNA,使β链合成减少。
2)β地中海贫血的临床分类 根据临床表现的严重程度的不同将β地中海贫血分为3种类型。
(1)重型β地中海贫血 又称Cooley贫血,患者是β0地中海贫血或β+地中海贫血的纯合子或β0地中海贫血与β+地中海贫血的双重杂合子,因β链生成受到抑制,以致含有β链的Hb A合成减少或消失,而多余的α链则与γ链结合而成为Hb F(α2γ2),使Hb F含量明显升高。由于Hb F的氧亲和力高,导致患者组织缺氧。过剩的α链形成包涵体附着于红细胞膜上而使其变硬,在骨髓内大多被破坏而导致“无效造血”。包涵体还会影响红细胞膜的通透性,从而导致红细胞的寿命缩短。由于以上原因,患儿在临床上呈慢性溶血性贫血,表现为面色苍白,肝、脾肿大,发育不良,常伴有轻度黄疸,症状随年龄增长而日益明显。贫血和缺氧刺激红细胞生成素的分泌量增加,促使骨髓增加造血,因而引起骨骼的改变,形成地中海贫血特殊面容,表现为头颅变大、额部隆起、颧高、鼻梁塌陷、眼距增宽。本病如不治疗,患者多于10岁前死亡。
(2)中间型β地中海贫血 患者是一些β+地中海贫血的双重杂合子和某些地中海贫血的变异型的纯合子,或两种不同变异型珠蛋白生成障碍性贫血的双重杂合子,其临床表现的轻重介于重型和轻型β地中海贫血之间,表现为中度贫血,脾脏轻度或中度肿大,黄疸可有可无,骨骼改变较轻。
(3)轻型β地中海贫血 患者是β0地中海贫血、β+地中海贫血或δβ地中海贫血的杂合子。由于β链的合成仅轻度减少,患者无症状或有轻度贫血,脾不肿大或有轻度肿大。本病容易被忽略,多在对重型患者家族调查时被发现。
三、血友病
血友病(hemophilia)是指一组由于血液中某些凝血因子的缺乏而导致严重凝血障碍的遗传性出血性疾病,男、女性均可发病,但患者大部分为男性。其主要类型有:血友病A(甲型)、血友病B(乙型)、血友病C(丙型),以及后来又发现的血管性假血友病。调查显示,我国总患病率约为2.73/100 000,其中男性患病率约为5.21/100 000,女性患病率约为0.06/100 000。各型的构成比例是:血友病A占79.8%,血友病B占14.1%,血友病C占2.8%,血管性假血友病占3.3%。在先天性出血性疾病中该病最为常见,出血也是该病的主要临床表现。较多见的是血友病A和血友病B。
1.血友病A(hemophilia A) 血友病A又称抗血友病球蛋白(antihemophilic globulin,AHG)缺乏症或第Ⅷ凝血因子缺乏症,主要表现为出血倾向,且出血特点为:①反复自发性缓慢持续渗血;②轻微创伤即可引发出血;③出血范围广泛。关节腔多次出血,可形成血肿,导致关节变形,颅内出血可造成死亡。实验室凝血检查见凝血时间延长,部分凝血活酶时间明显延长。血浆抗血友病球蛋白合成减少或完全缺乏。
本病为X连锁隐性遗传,故男性患者较多。该病在男性中发病率约为1/5 000。女性杂合子为携带者。其基因定位在Xq27,全长186kb,是人类最大的基因之一,包括26个外显子和25个内含子。
经研究表明,本病近1/3的患者无家族史,而是由AHG基因自发突变形成的。世界已报道的相关突变达900多种,突变类型包括点突变、缺失、插入和倒位。内含子22倒位是重型血友病A致病的主要原因。携带者的检出对预防该病患儿的出生很重要。可以应用Southern印迹杂交及PCR技术等进行诊断或进行产前诊断。
2.血友病B(hemophilia B) 血友病B也称血浆凝血活酶成分(plasma thromboplastic component,PTC)缺乏症或第Ⅸ凝血因子缺乏症,遗传方式也是Ⅹ连锁隐性遗传,患者的临床表现与血友病A相似,但发病率较低。由于杂合子的第Ⅸ凝血因子的活性仅为正常值的1/3左右,某些女性杂合子可出现症状,故该病的女性患者较血友病A的多见。
人类第Ⅸ凝血因子基因定位于Xq27.1,长度为34kb,由8个外显子和7个内含子组成。由其编码的血浆凝血活酶成分由415个氨基酸组成。基因突变类型较多,涉及单碱基置换、缺失、插入和移码突变,大部分是单碱基置换。
3.血友病C(hemophilia C) 血友病C又称血浆凝血活酶前质(plasma thromboplastic antecedent,PTA)缺乏症或第Ⅺ凝血因子缺乏症。该病的症状通常比血友病A、血友病B的轻,发生在肌肉及关节的血肿和积血比较少见。与其他血友病相比,血友病C的发病率较低,仅为血友病A的1/50左右,且有明显的种族发病倾向,多见于土耳其南部犹太人后裔。遗传方式属常染色体隐性遗传。基因定位于4q35,基因长度为23kb,包括15个外显子和14个内含子,可编码625个氨基酸。
4.血管性假血友病(von Willebrand disease) 血管性血友病因子(vWF)是一种大分子血浆糖蛋白,分布于血浆、血小板及内皮细胞下的血管基质。其功能表现为与第Ⅷ凝血因子结合并保护其免受未成熟蛋白溶酶的灭活。vWF基因定位于12p 13.3,基因长度为178kb,有52个外显子,可编码2 813个氨基酸。vWF的缺乏常伴有第Ⅷ凝血因子的活性降低,本病患者有明显的出血倾向,形成血管性假血友病。病情不如血友病A的严重。
四、假肥大型肌营养不良症
假肥大型肌营养不良症是指由于基因突变影响了抗肌萎缩蛋白在横纹肌组织中的表达而引发的一种常见的遗传性肌病,为X连锁隐性遗传病,包括杜氏进行性肌营养不良和贝克型进行性肌营养不良两种类型。
1.杜氏进行性肌营养不良(DMD) 该病由Duchenne于1868年首先描述,并由此命名。患儿多为男性,在活产男婴中发病率约为1/3 500,地理或种族间的差异不明显。患儿主要临床表现为:行走较慢,不能正常跑步,容易跌倒,因肢体近端骨骼肌进行性萎缩无力而导致走路时向两侧摇摆,呈典型鸭步。由仰卧转为站立时,患儿须先转为俯卧位,然后以双手支撑足背、膝部等部位,方能站立。一般9~12岁时患儿不能独立行走。多数患儿心肌受累,表现为窦性心动过速。随着年龄增长,病情加重,可出现心脏增大、呼吸衰竭,存活时间很少超过30岁。患者病情的严重程度与该病在家族中遗传代数成反比,即受累代数越多,病情越轻。
DMD基因定位于Xp21,是人类最大的基因之一,总长2.5Mb,占全部基因组序列的0.1%,占X染色体全长的1.5%。该基因组序列主要由内含子组成,包括79个外显子和78个内含子。基因突变方式主要是缺失,还有点突变和微缺失。
2.贝克型进行性肌营养不良(BMD) 该病与DMD是由同一种基因引起的疾病,发病率比DMD的低,与DMD相比该病患者发病年龄较晚,症状较轻,多不伴有心肌受累或仅有轻度受累,病情进展缓慢。患者多在20岁左右还能行走,寿命可达40~50岁。
五、家族性高胆固醇血症
家族性高胆固醇血症(familial hypercholeslerolemia,FH)是一种受体蛋白病,受体是存在于细胞膜上、细胞质中或细胞核内的一类具有特殊功能的蛋白质。现已发现包括多肽类激素受体、固醇类激素受体以及脂蛋白受体在内的具有调节生理功能的特异性受体达30种以上。受体的本质是蛋白质,基因突变可使受体的生物功能异常、合成数量异常或结构异常,从而影响代谢过程的正常进行。这种由受体蛋白遗传性缺陷引起的疾病,称为受体蛋白病(receptor protein disease)。
家族性高胆固醇血症是由于细胞膜上低密度脂蛋白(LDL)受体缺乏所导致的疾病。在正常代谢过程中,LDL先与LDL受体结合,然后通过内吞作用进入细胞,被溶酶体内的酸性水解酶水解。水解后释放出的游离胆固醇可激活脂酰辅酶A:胆固醇脂酰转移酶(acyl-CoA:cholesterol acyltransferase,ACAT),本身被酯化成胆固醇脂而储存。游离的胆固醇还可抑制β-羟基-β-甲基戊二酰辅酶A(HMG CoA)还原酶的生物活性,从而减少胆固醇的合成。成纤维细胞低密度脂蛋白受体作用示意图如图8-4所示。该病的发病机制是LDL受体基因发生突变,突变方式包括缺失突变、错义突变、无义突变、整码突变及移码突变等多种类型。由此引起LDL受体功能异常,主要有4种表现:①LDL受体合成的数量减少;②LDL受体转运不良,是指不能将LDL受体从粗面内质网运送到高尔基复合体;③LDL受体与LDL不能结合或结合能力下降;④LDL受体与LDL结合后向细胞内移的功能异常。
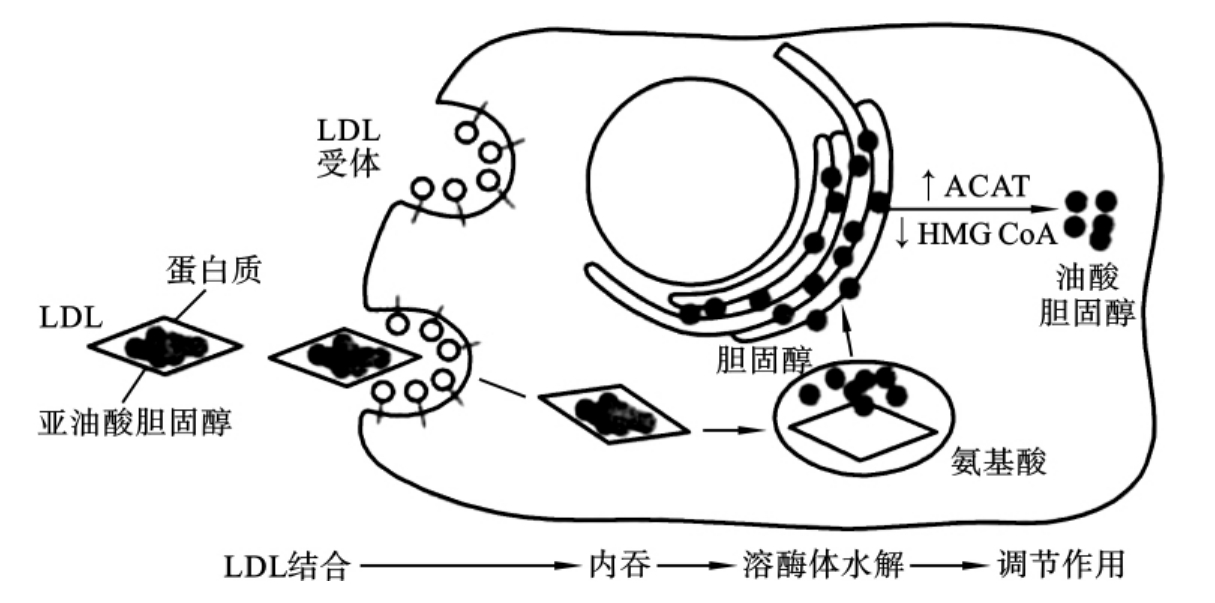
图8-4 成纤维细胞低密度脂蛋白受体作用示意图
本病为常染色体不完全显性遗传,患者多为杂合子,患病率约为1/500。LDL基因定位于19p13,为单一序列基因,总长45kb,包括18个外显子和17个内含子。患者因细胞膜上的LDL受体缺陷,LDL进入细胞数量减少,细胞内胆固醇的抑制作用受阻,使细胞内胆固醇合成升高,因胆固醇沉积而出现黄色瘤,并随年龄增长而日益严重。
(关丽娜)

