
高血糖症是指血液中葡萄糖的浓度长期高于正常水平。
判断标准:空腹血糖水平高于6.9mmol/L及餐后2小时血糖高于11.1mmol/L。
当血糖超过其肾阈值9.0mmol/L时,出现尿糖。
高血糖包括生理性高血糖和病理性高血糖。
一、病因与发病机制
高血糖症主要由机体胰岛素绝对/相对不足或利用低下引起。
1. 胰岛素绝对不足
胰岛素绝对不足主要是指胰岛B细胞损伤,导致胰岛素生成或分泌障碍,使血液中胰岛素含量降低。
(1)免疫因素
胰岛素生成或分泌不足的关键环节是胰岛B细胞的进行性损害。
细胞免疫异常:包括介导细胞毒性T淋巴细胞,针对胰岛B细胞特殊抗原产生的破坏作用;激活的T淋巴细胞、巨噬细胞、粒细胞释放多种细胞因子,在胰岛B细胞自身免疫损伤中起重要作用。
自身抗体形成:胰岛细胞自身抗体的产生与B细胞的损伤有关。
(2)胰岛素基因突变
胰岛素基因表达的任何环节出现障碍,如胰岛素基因点突变,可引起一级结构的改变,C肽裂解点的氨基酸不正常,可使胰岛素原转变成胰岛素不完全,变异胰岛素与受体结合能力或生物活性降低甚至丧失。
(3)遗传因素
遗传易感性在胰岛素分泌障碍发生中起重要作用,某些相关的基因突变可促发或加重胰岛B细胞自身免疫性损伤过程。
组织相容性抗原基因:位于6号染色体上的组织相容性抗原(HLA)基因可引起胰岛素分泌障碍。HLA-Ⅱ类抗原与胰岛B细胞自身免疫性损伤密切相关。胰岛B细胞免疫耐受性选择性丧失,可使其易于受到环境因素与特殊细胞膜抗原的相互作用的影响,进而发生自身免疫性损伤。
细胞毒性T淋巴细胞的相关性抗原4基因(CTLA-4):CTLA-4 49/A的多态性表达,可以激活各种T淋巴细胞,导致胰岛B细胞自身免疫反应性破坏。
Fox基因:FoxP 3表达异常,CD4+CD25+Treg细胞减少,不足以维持自身免疫耐受,经由T细胞介导可引起胰岛B细胞选择性破坏。
胸腺胰岛素基因表达:胸腺胰岛素基因表达异常,影响胸腺对胰岛素反应性T细胞的选择,与HLA-Ⅱ协同作用导致胰岛B细胞破坏。
(4)胰岛B细胞凋亡
凋亡是胰岛B细胞损害的严重机制,其作用途径:
通过Fas-FasL途径:Fas/FasL及相关因子组成Fas系统,在传递细胞凋亡信号、发挥免疫监控中起重要作用。
磷脂酶A2(PLA2)激活:激活的PLA2可诱导胰岛B细胞凋亡。
细胞因子:白细胞介素-1β(IL-1β)、大鼠干扰素-α(INF-α)和干扰素-γ(IFN-γ)通过诱导胰岛B细胞凋亡而损害胰岛B细胞。细胞因子在诱导胰岛B细胞凋亡的过程中具有协同作用。
(5)环境因素
引起胰岛B细胞损坏的有关环境因素主要有病毒、化学因素、饮食因素等,以病毒感染最为重要。
病毒感染:胰岛B细胞损伤机制是, 1)病毒直接破坏B细胞;2)病毒作用于免疫系统,诱发自身免疫;3)分子模拟作用使胰岛细胞失去免疫耐受 ;4)刺激调节性T细胞及效应细胞,引起胰岛B细胞的自身免疫损伤。
化学损伤:四氧嘧啶等对胰岛B细胞有毒性作用的化学物质或药物,对胰岛细胞有直接毒性作用;或通过化学物质中的-SH直接导致胰岛B细胞溶解,并可诱导胰岛B细胞产生自身免疫,导致胰岛B细胞进一步损伤,数量减少。
饮食因素:例如牛奶蛋白与B细胞表面抗原交叉免疫反应,导致胰岛B细胞的自身免疫性损害。
2. 胰岛素相对不足
胰岛素相对不足主要由胰岛素抵抗引起。
胰岛素抵抗(insulin resistance,IR)是指胰岛素作用的靶组织和靶器官(主要是肝脏、肌肉和脂肪组织)对胰岛素生物作用的敏感性降低,可引起高血糖症,而血液中胰岛素含量可正常或高于正常。
胰岛素抵抗的发病与遗传缺陷高度相关,根据这种缺陷相对于胰岛素受体的位置,可分为受体前、受体和受体后三个水平。
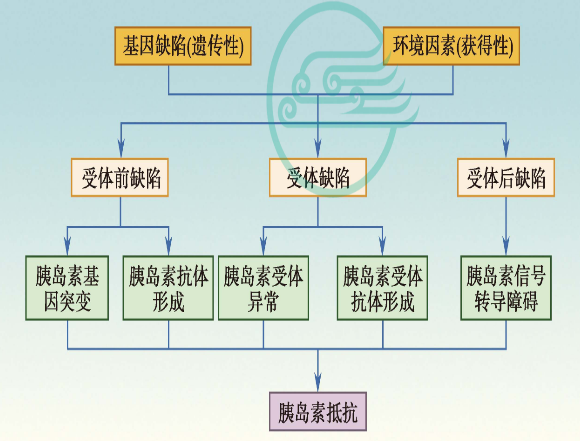
(1)受体前缺陷
胰岛素生物活性下降,失去对受体的正常生物作用。
最常见的原因是胰岛素抗体生成,包括内源性抗体和外源性抗体。
(2)受体缺陷
受体缺陷是指细胞膜上胰岛素受体(insulin receptor,Ins R)功能下降或数量减少,胰岛素不能与受体正常结合,使胰岛素不能发挥降低血糖的作用。
InsR水平的胰岛素抵抗产生主要包括InsR功能和结构异常。
InsR异常:多由胰岛素受体基因(IRG)突变引起,导致受体数量减少或活性降低。
InsR抗体形成:胰岛素受体抗体(insulin receptor antibodies,IRA)不仅可与细胞膜上胰岛素受体结合,使细胞表面的受体数量减少,还可竞争性抑制胰岛素与其受体的结合,导致受体后的信号转导发生障碍。
(3)InsR后水平的缺陷
胰岛素与靶细胞受体结合后,信号向细胞内传传导发生异常所引起的一系列代谢过程,包括信号传递、放大、蛋白质-蛋白质交联反应,磷酸化与脱磷酸化以及酶促级联反应的异常,从而产生胰岛素抵抗。
胰岛素信号转导障碍是产生胰岛素抵抗和高血糖症的主要发生机制,但其中许多机制目前尚未完全明确。
胰岛素信号转导主要途径——磷酸肌醇-3-激酶(PI3-K)转导途径的步骤:
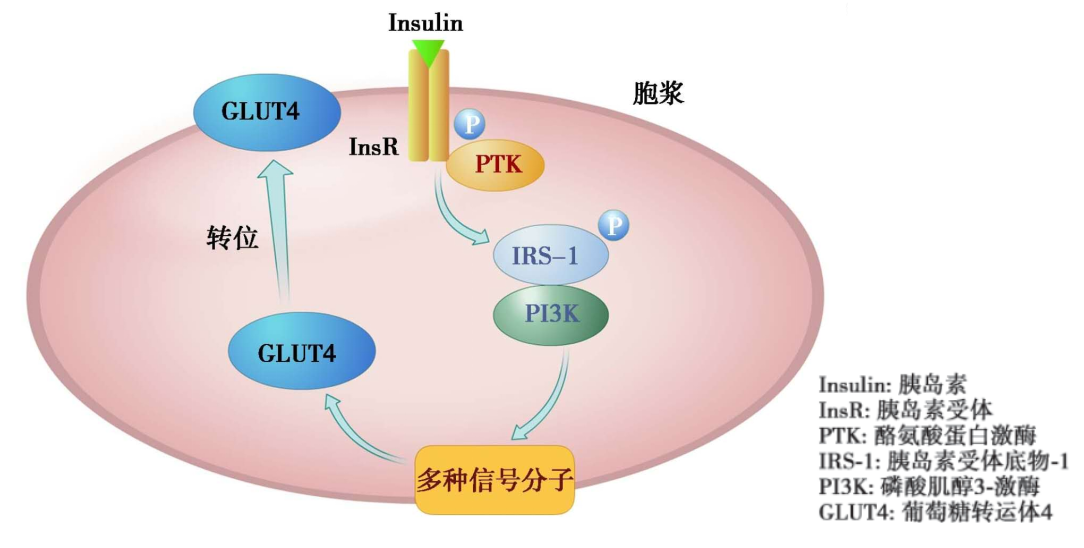
胰岛素信号转导异常主要发生在以下水平:
1)IRS异常,包括IRS的不正常降解 、IRS磷酸化异常 、IRS的分布异常 ;
2)PI3K异常
3)PKB异常
4)GSK-3异常
5)GLUT4异常
3. 胰岛素拮抗激素失调
胰高血糖素(glucagon)是维持血糖稳态的关键性调节激素。血糖浓度负反馈调节胰高血糖素的分泌。
高胰高血糖素血症所致的肝糖原分解和糖异生过多,是高血糖发病的重要环节。
(1) 胰高血糖素分泌的抑制受损
(2)胰岛A细胞对葡萄糖的敏感性下降
(3) 胰高血糖素对B细胞的作用异常
(4)胰岛A细胞的胰岛素抵抗
4. 其他因素
(1)肝源性高血糖 肝硬化、急慢性肝炎、脂肪肝等,可引起糖耐量减退,脂肪肝等
(2)肾源性高血糖 尿毒症、肾小球硬化等肾功能严重障碍
(3)应激性高血糖 外科手术、严重感染、烧伤、休克等
(4)内分泌性高血糖 肢端肥大症、嗜铬细胞瘤、甲亢等
(5)妊娠性高血糖
(6)药物性高血糖
(7)其他因素引起的高血糖 肥胖、高脂血症、某些肌损伤及遗传病、有机磷中毒等
二、高血糖症对机体的影响
1. 代谢紊乱
胰岛B细胞胰岛素分泌能力和(或)胰岛素生物作用缺陷,可导致胰岛素绝对或相对不足,引起一系列代谢紊乱
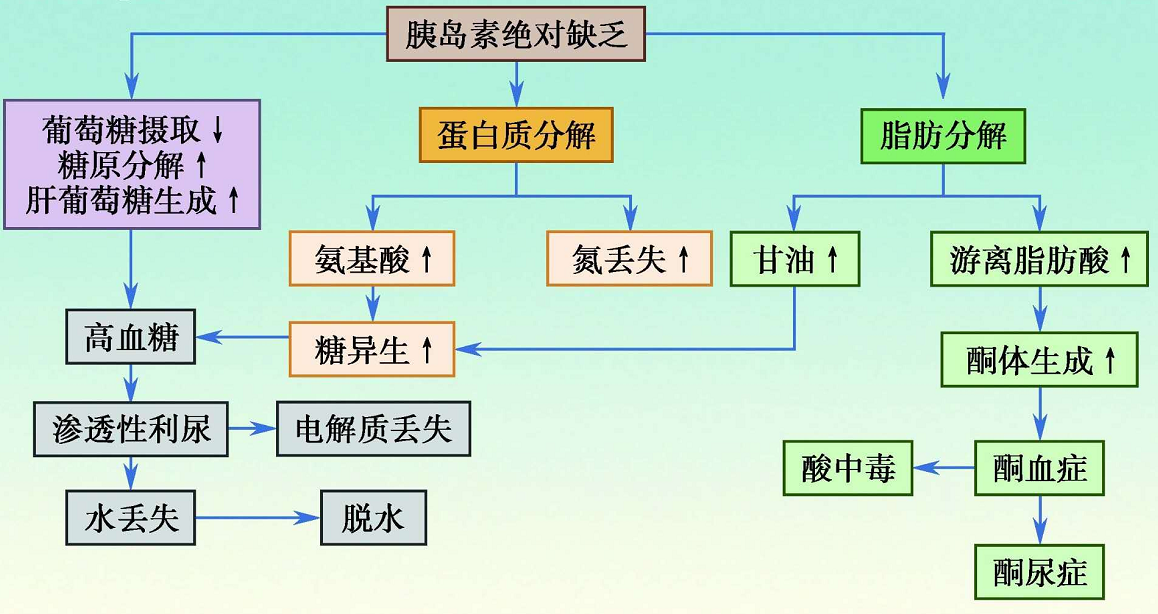
(1)渗透性脱水和糖尿
①血糖急剧升高引起细胞外液渗透压增高,水从细胞内转移至细胞外,可导致细胞内液减少,引起细胞脱水。脑细胞脱水可引起高渗性非酮症糖尿病患者昏迷。
②血糖浓度高于肾糖阈,肾小球滤过的葡萄糖多于肾小管重吸收的葡萄糖,葡萄糖在肾小管液中的浓度升高,小管液中的渗透压明显増高,阻止了肾小管对水的重吸收,丢失大量的细胞外液,从而出现渗透性利尿和脱水,临床表现为糖尿、多尿、口渴。
(2)物质代谢紊乱
胰岛素分泌绝对不足和(或)胰岛素生物学效应降低,肝脏、骨骼肌、脂肪组织等效应器官,对葡萄糖的摄取、利用减少,肝糖原分解加强,引起高血糖;脂肪组织从血液摄取甘油三酯减少,脂肪合成降低;脂蛋白酯酶活性降低,血游离脂肪酸和甘油三酯浓度升高;蛋白质合成减少,分解加速,出现负氮平衡。
(3)酮症酸中毒
高血糖症时,由于机体不能很好地利用血液中的葡萄糖,各组织细胞处于糖和能量的饥饿状态,可引起脂肪分解加速,血中游离脂肪酸增加,酮体生成增加超过了酮体的利用,大量酮体堆积在体内形成酮症,发展为酮症酸中毒和高钾血症。
2. 多系统器官损害
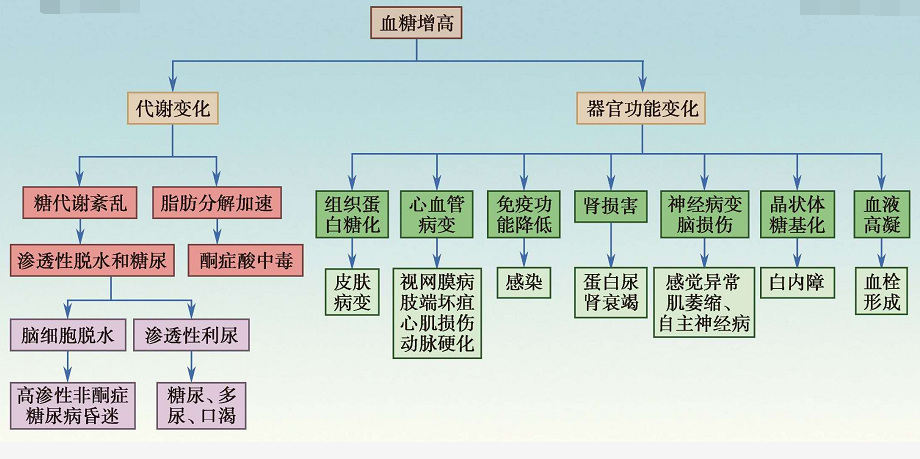
(1)心血管系统病变
高血糖引起的微血管典型改变是微循环障碍和微血管基底膜增厚,病变主要表现在视网膜、肾、神经和心肌组织,其中尤以高血糖肾病和视网膜病最为重要;而大血管病变可导致动脉粥样硬化的发生,主要侵犯主动脉、冠状动脉、脑动脉、肾动脉和肢体外周动脉等,引起冠心病、缺血性或出血性脑血管病、肾动脉硬化、肢体动脉硬化等。
(2)神经系统病变
高血糖所引起的神经病变,包括外周神经病变和自主神经病变,其发生机制可能与高血糖所致的代谢或渗透压张力的改变有关。
高血糖是急性脑损伤的促发因素之一,它在导致脑缺血的同时还可继发神经元的损伤、增加脑血管意外的概率。
(3)免疫系统病变
高血糖对免疫系统的影响主要表现为使吞噬细胞的功能降低。
其发生机制是:①高血糖减弱中性粒细胞和单核细胞的黏附、趋化、吞噬和杀菌作用。②高血糖可升高血中超氧化物浓度和硝基酪氨酸(NT)的水平。
血糖增高易发生念珠菌及其他一些罕见的感染;长期尿糖阳性的女性易发生阴道炎。
(4)血液系统病变
高血糖可引起血液系统凝固性增高,导致血栓形成。
发生机制:纤维蛋白溶解酶原激活物抑制剂活性增加;纤维蛋白溶解酶原激活物活性降低;全血黏度和血浆黏度增高;血液高渗,血黏度的升高。
当血浆黏度增高时,血流量减少,不利于组织灌流,造成组织缺血,易造成血栓性疾病,这是临床上高血糖症合并冠心病及其他慢性血管病变的重要病理基础之一。
(5)晶状体病变
长期高血糖,可导致晶状体肿胀,出现空泡,某些透明蛋白变性、聚合、沉淀,导致白内障。
(6)肾脏病变
长期高血糖,通过改变肾脏血流动力学以及代谢异常引起的肾脏功能病变。
主要表现为:蛋白尿、水肿、电解质平衡紊乱、高血压和氮质血症等。
(7)肢端坏疽
主要表现为进行性肢端缺血、手足麻木及溃烂坏死。
主要原因是肢端缺血、缺氧、水肿、营养物质匮乏、代谢产物堆积,细菌容易感染而发生干性坏疽。
病理生理基础是血管病变、周围神经病变合并感染。
(8)高血糖对其他器官、系统的影响
高血糖时,由于组织蛋白糖基化作用( glycosylation)増加以及血管病变,皮肤出现萎缩性棕色斑、皮疹样黄瘤。
长期血糖增高所引起的代谢紊乱、血管病变,可导致骨和关节的病变,如关节活动障碍、骨质疏松等。
3. 高血糖防治的病理生理基础
(1)饮食治疗
(2)运动疗法
(3)药物治疗
降糖药物
口服降糖药物包括增加胰岛素敏感性或刺激胰岛素分泌的药物。如磺脲类药物格列本脲、格列吡嗪、格列奇特等,主要作用是刺激胰岛B细胞分泌胰岛素,其作用部位是胰岛B细胞膜上的ATP敏感钾离子通道(ATP- sensitive potassium channel,KATP)。
胰岛素治疗
应用外源性的胰岛素可快速有效地降低血糖浓度,控制高血糖症;或作为体内胰岛素绝对缺乏的终生替代治疗,有可能延缓自身免疫对B细胞的损害。
在使用降糖药物尤其是胰岛素时,应密切监测患者血糖水平,防止因剂量过大而导致低血糖反应。由胰岛素用量过大引起的低血糖,严重时可因中枢神经系统的代谢被抑制引起昏迷和休克,即胰岛素休克( insulin shock)。
其他治疗
可进行胰腺移植、胰岛细胞移植、干细胞治疗等,以替代损伤的胰岛B细胞分泌胰岛素。

