-
1 内容
-
2 作业
儿科遗传性疾病
Francis Collins:
“Except for some cases of trauma, it is fair to say that virtually every human illness has a hereditary component.”
除某些外伤之外,实践上可以说每个人类的疾病都有遗传因素

Collins FS (1999) Shattuck Lecture: medical and societal consequences of the Human Genome Project. N Engl J Med. 1999; 341:28-37
基因的作用
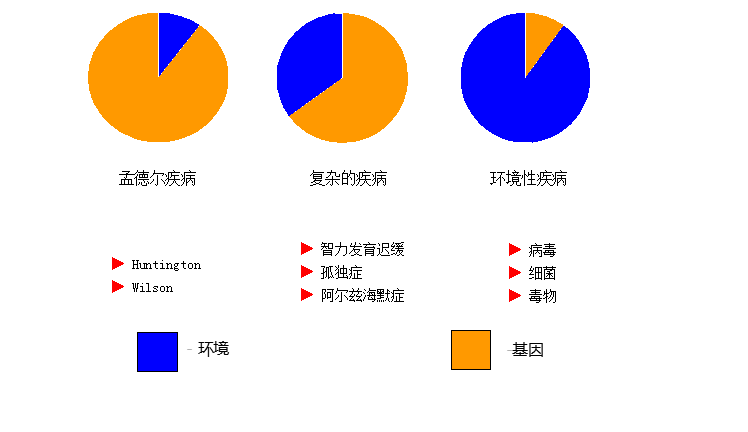
遗传病分类
◇ 染色体病(Chromosomal Abnormalities)
21 三体综合征
◇ 单基因病(Single Gene Defects)
苯丙酮尿症(PKU)
http://www.ncbi.nlm.nih.gov/Omim
◇ 多基因病(Multifactorial Disorders)
◇ 线粒体病 (Mitochondrial disease)
◇ 基因组印记(Genomic imprinting)
Number of Entries in OMIM (Updated 28032015)


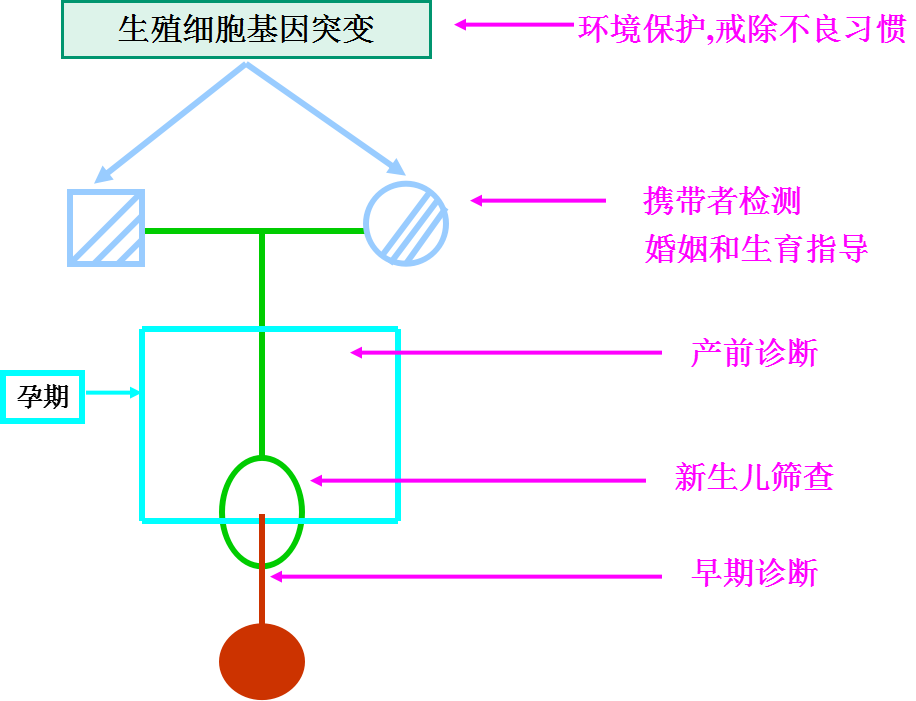
新生儿:遗传病“重灾区”
近7000种,
▪ 目前诊断困难、治疗手段有限
▪ 大多数疾病分子机制和病理生理机制不明
▪ 新的基因检测技术为病因学诊断提供条件
▪ 临床转化应用

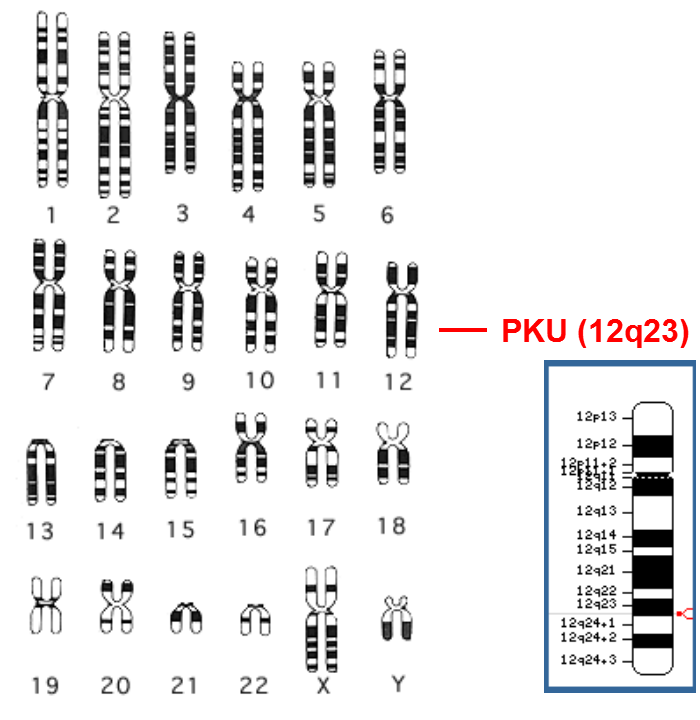
苯丙酮尿症
(Phenylketonuria-PKU)
▪ 1934,首次报道
▪ AR,基因于染色体12
▪ 1/ 60携带者
▪ 发病率:
- 1/10,000 (美国)
- 1/16,5000 (中国)
▪ 苯丙氨酸高,未治疗智能低下

Molly’s Story
发病机制
Each gene occurs at a specific site, or locus, on a chromosome.
Metabolism basics
▪ Metabolism is the sum total of all chemical reactions in the body
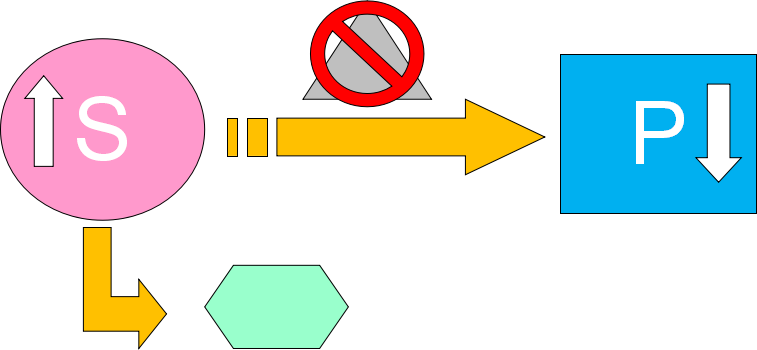
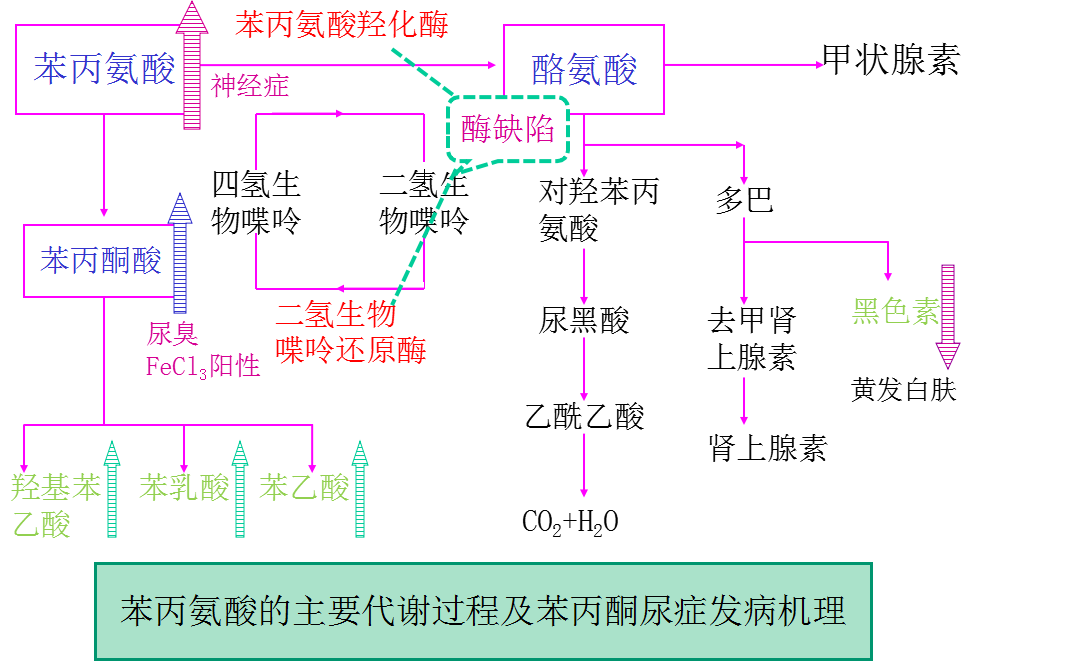
苯丙氨酸代谢过程
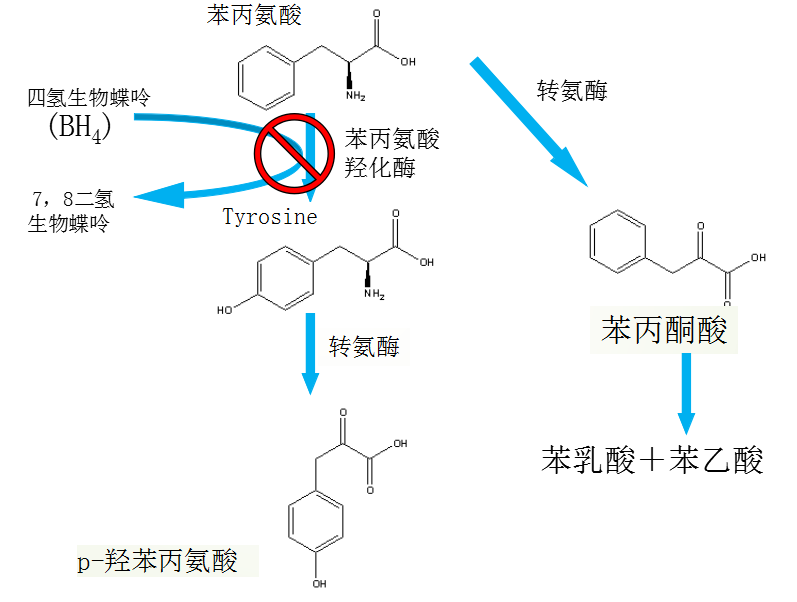
PAH mutations
▪ Most common mutations (North America)
- R408W Classical PKU (18.7%)
- IVS12G-A+1 Classical PKU (7.8%)
- Y414C Mild hyperphe (5.4%)
- 13 other mutations (1-5%)
- 55 other mutations (<1%)
点突变导致苯丙氨酸羟化酶(PAH )缺陷
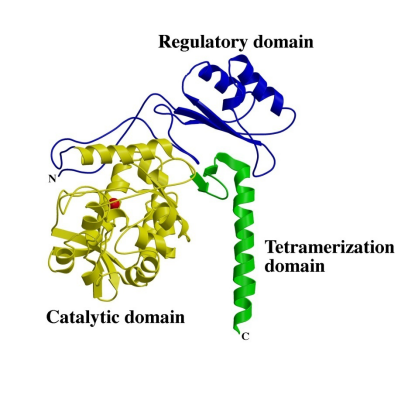
– PAH单体3D 结构
– PAH 四聚体结晶
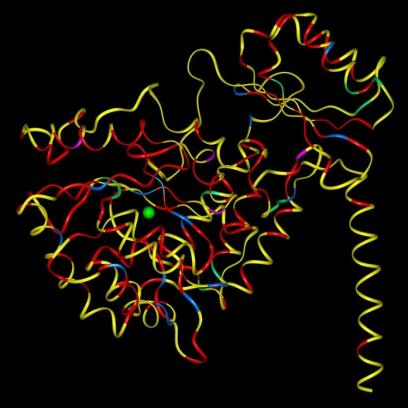
–红、蓝、绿示点突变
– 黄无突变
临床表现
Clinical - Genetic heterogeneity
▪ Classical PKU
- “severe”
- <1% residual enzyme activity
- very high levels PHE-strict diet for life
▪ Type II/Atypical PKU
- milder
- tolerate more liberal protein diet
▪ Type III/Mild/Benign persistent Hyperphe
- 5% residual activity
- levels <600uM
- no diet needed
▪ Type IV/Malignant PKU 2%
- BH4 cofactor defect
- need neurotransmitter replacement therapy
- outcome often not good
未处理表现
▪ 早期症状:见于 50%
- 呕吐、激若、湿疹性皮炎、鼠尿
▪ 晚发症状:
- 神经系统症状
- 严重脑部问题
▪ 其他常见特征:
- 小头,突出面颊和上颌,齿稀疏、釉质发育不全,生长落后
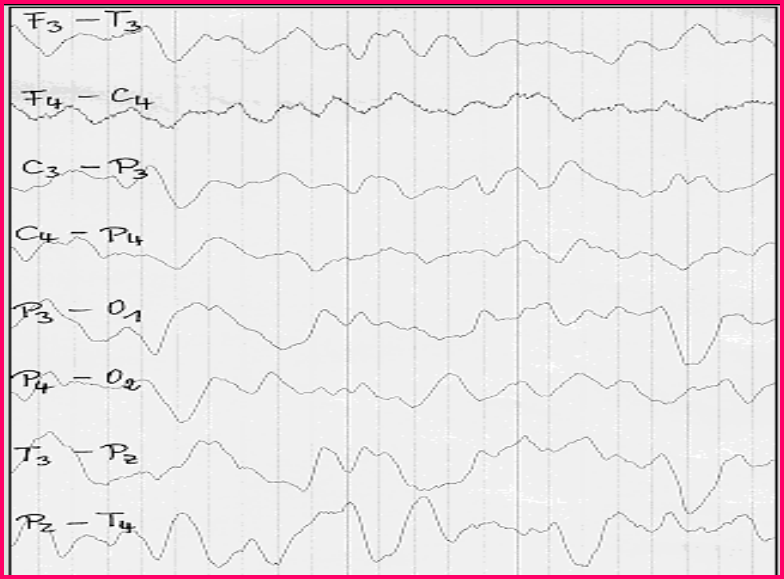
8月龄苯丙酮尿症脑电图改变:轻度弥漫性脑功能障碍
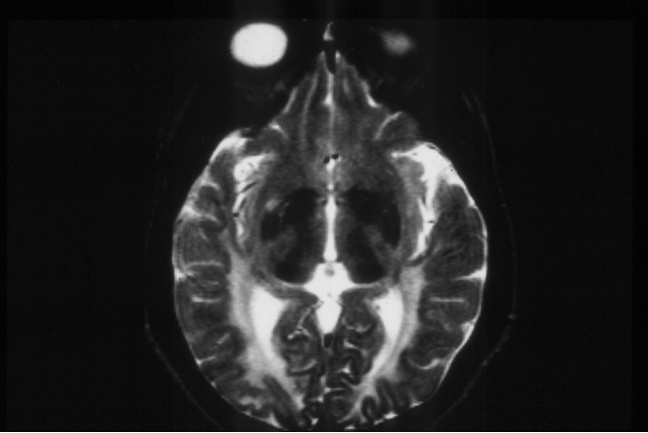
PKU MRI - abnormal white matter
Even in treated PKU there are neurological consequences-
learning, executive function problems etc
经典型 PKU 的诊断依据为
(1)母乳或规则喂食配方乳的婴儿苯丙氨酸血浓度为≥1200μmol/l(20 mg/dl);
(2)酪氨酸血浓度 < 250μmol/l(5 mg/dl)和
(3)诊断 BH4 所需的尿喋啉指标值正常
苯丙氨酸胚胎病 (所谓的苯丙酮尿症综合征)
▪ 低出生体重(low birth weight)
▪ 精神运动发育落后(psychomotor-retardation)
▪ 小颅畸形(microcephaly)
▪ 先天性胚胎病(congenital embryopathy)
未控制PKU风险
首次报道 Dent (1957), Marby (1963)
▪ 产前:
- 流产 (24%)
- IUGR (40%)
- 心理问题(92%)
- 先心(10%)
▪ 产后:
- 发育迟滞、神经异常、颅面部畸形
- 小头 (73%)
苯丙酮尿症的实验室检查
▪ 三氯化铁试验(尿):生后4-6周,假阳性率高
▪ Guthrie枯草杆菌抑制试验:新生儿筛查
▪ 血清苯丙氨酸浓度测定:明确诊断
▪ 四氢生物喋呤负荷试验:鉴别各型苯丙酮尿症
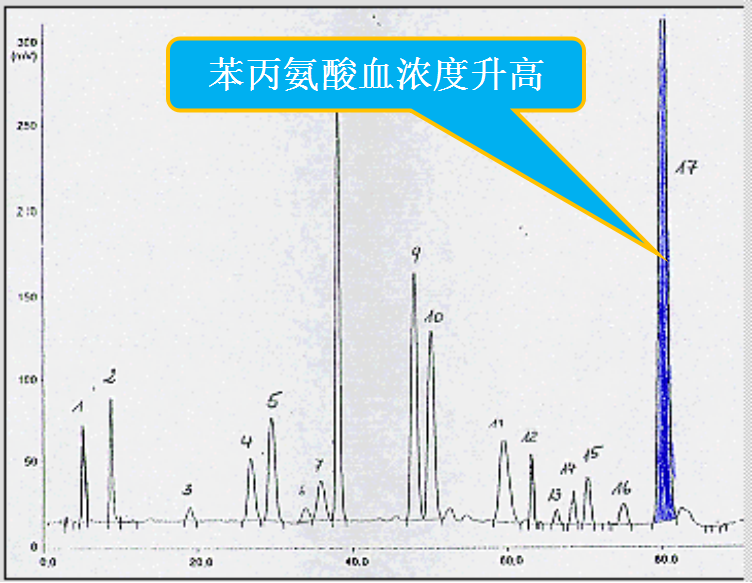
氨基酸分析仪

正常血苯丙氨酸浓度
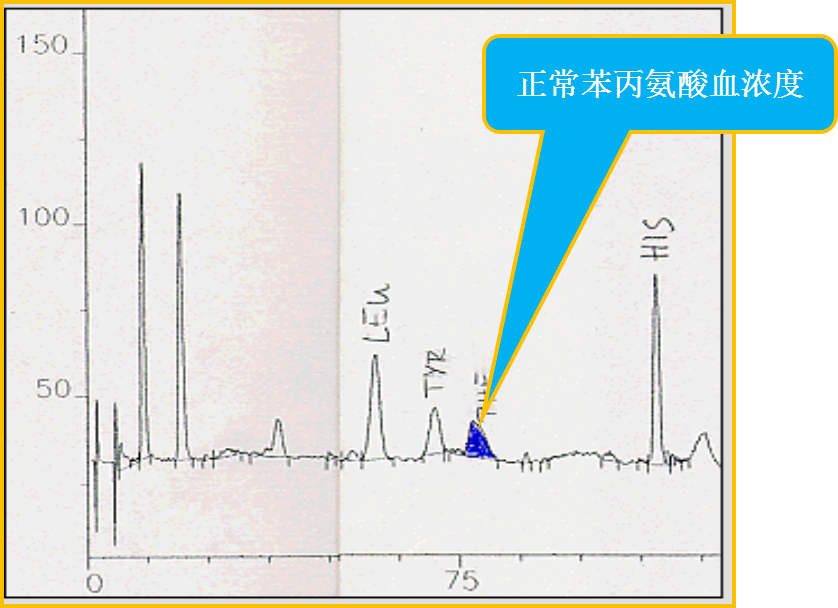
▪ BH4 负荷试验
- 给予BH4 后测定血尿样本,若Phe and Tyr 仍高水平,可诊断经典 PKU
▪ 串联质谱:
- 筛选 >40 氨基酸、有机酸和脂肪酸氧化疾病包括经典 PKU,高苯丙氨酸血和生物蝶呤辅酶缺陷
- 测定Phe/Tyr比率,降低假阳性
预防和治疗
新生儿PKU筛查出血苯丙氨酸血浓度升高者的随访步骤
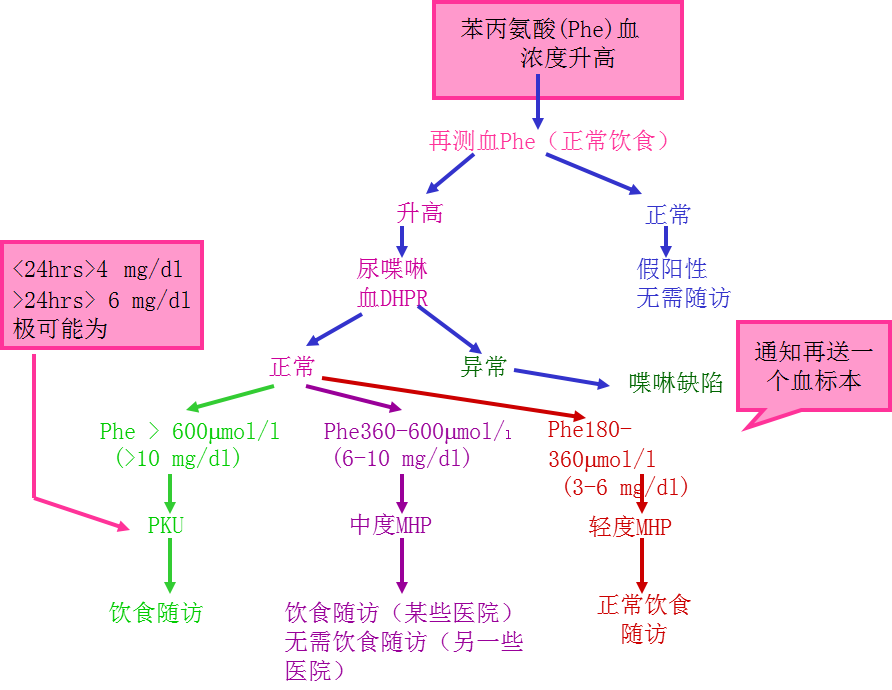
对所有苯丙氨酸血浓度持续在360μmol/l(6mg/dl)以上者于生后 20 天内即开始饮食治疗

母亲苯丙氨酸血浓度 > 1,200μmol/l(20 mg/dl)者的子女 95%将有智能迟滞

苯丙酮尿症患者的食物
▪ 主要食用含苯丙氨酸浓度低的食物
- 多种蔬菜和水果(苹果,梨,西瓜,生菜,黄瓜.番茄,胡萝卜,南 瓜等)
- 黄油,植物油,油
- 果酱,果子酱,蜂蜜糖浆
- 含苯丙氨酸少的面包,面条,饼干和米饭,米粉
- 汽水,可乐.茶,咖啡,矿泉水
按对苯丙氨酸耐受情况应予以避免的食物
▪ 肉,香肠,鱼
▪ 蛋
▪ 奶,酸奶,奶酪,凝乳
▪ 核桃
▪ 巧克力
▪ 含奶的冰淇淋
▪ 含天冬氨酸的奶制品
治疗时间:
为避免MR,最好在生后60天内开始. 建议终身治疗。
70 例在7岁停止PKU饮食,IQ降低, 32% vs 78%大学毕业
phe水平监测:
每月抽血,安全 phe 水平 2- 6 mg/dL.
记录phe摄入量,根据血phe调整
苯丙酮尿症的预防