
-
1 内容
-
2 作业
先天性肾上腺皮质增生症
概述
◇ CAH: 是一组因肾上腺皮质激素合成途径中酶缺陷引起的常染色体隐性遗传病
◇ 新生儿发病率: 约1/100001/15000。
◇ 常见的CAH类型有:21-羟化酶缺乏、11β-羟化酶缺乏、17-羟化酶缺乏、类脂性肾上腺皮质增生症、3β-羟类固醇脱氢酶缺乏、18-羟化酶缺乏。
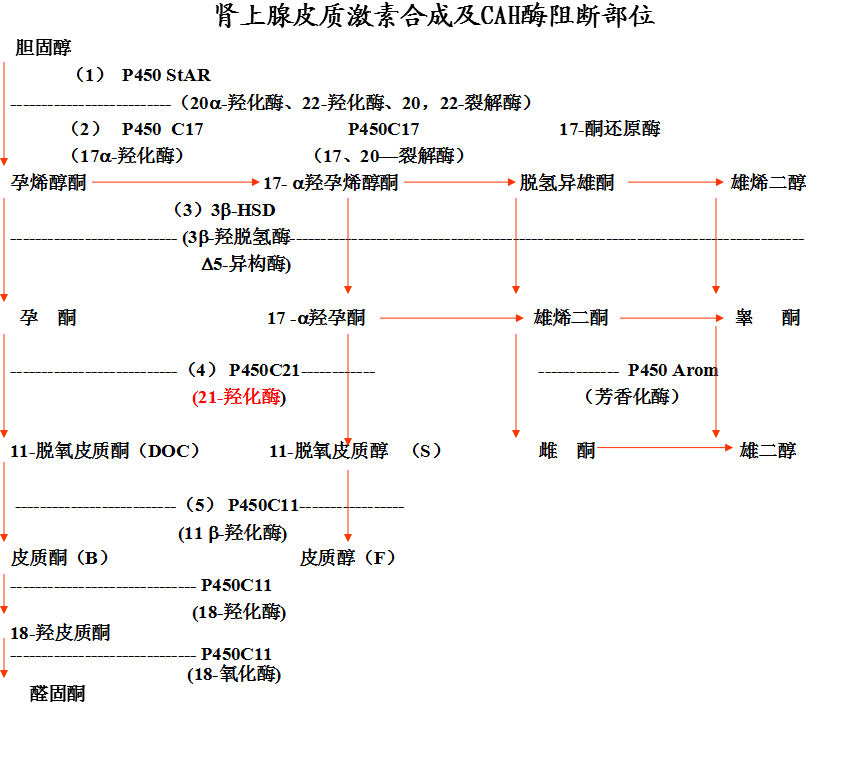
21-羟化酶缺乏
◇ 21-羟化酶缺乏者最为多见,95 %以上的CAH患儿为该酶缺乏所引起。
◇ 经典型 :75%失盐型, 25% 单纯雄性化
◇ 非典型性占CAH中的1/3
◇ 经典型 21-HD 15,000-20,000
遗传机制
◇ 体内存在二种 21-羟化酶基因, CYP21A1和CYP21A2,位于 6p21.3.
◇ CYP21A2是活性基因.
◇ CYP21A1与 CYP21 98% 等同,由于存在9个不同的突变为伪基因(pseudogene)
◇ 超过 90%的 21-HD基因突变是上述二种基因的不同重组
◇ 不同的基因突变导致不同程度的疾病
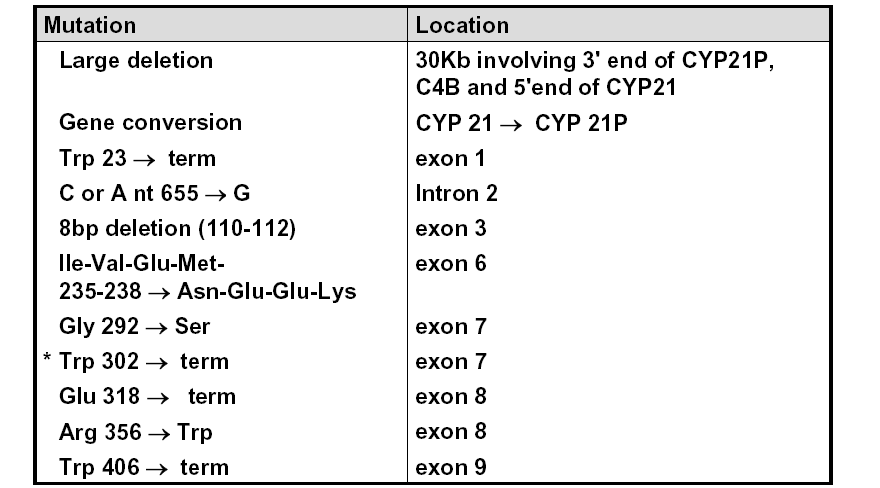
与21-羟化酶缺乏相关的严重突变

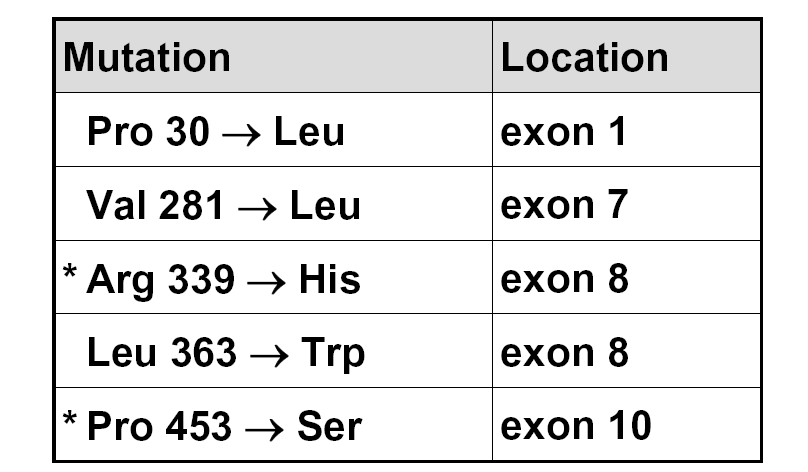
与21-羟化酶缺乏相关的微小突变
21-羟化酶缺乏CAH的常见临床表现
经典型21-羟化酶缺乏
◇ 特征:
√ 皮质醇 单纯男性化型可正常
√ PRA不同程度,SW 醛固酮早期,失代偿减少
√ 单纯男性化型大多正常
√ ACTH不同程度增加
√ 孕酮、17-OHP、DHEA、T合成增加
√ 尿17-酮皮质类固醇、类固酮,游离皮质醇
√ 醛固酮合成减少导致失盐症状:高钾低钠 ; 血浆肾素活性增加
√ 酶活性部分缺乏者,醛固酮合成不受影响,患者血钠钾正常
√ 过量的雄激素分泌导致女性雄性化,二性畸形,男孩阴囊皮肤色素成着
21-羟化酶缺乏
◇ 同一家庭的不同个体可以有经典型、非经典型和无症状型,其原因可能与等位基因的不同变异有关。
◇ 美国17-OHP质谱筛查
21-羟化酶缺乏-非典型性CAH
◇ 非典型性占CAH中的1/3
◇ CYP21A2活性为正常人的20%-50%
◇ 多见于女性,发生率有明显的种族差异,发生率在白种妇女中较高。
◇ 患者与出生时无临床症状,外生殖器正常,随着年龄增大,多数在儿童期或成年期逐渐出现雄激素增高的体征。男孩早期可出现胡须、阴毛、痤疮。
◇ 发病后身高增长快速,而其成年期最终身高常明显低于预测身高
◇ 成年期发病的男性因雄激素增高,其体征常被认为是正常的性发育,不宜察觉,多数会因精子减少而致生育功能障碍;
◇ 女性患者为高雄激素血症,除多毛外,月经初潮延迟、还会出现继发性月经过少或闭经。
◇ 部分患者是混合轻微的和几个CYP21A2突变的杂合体
◇ 月经量少、多(粗)毛, 多囊卵巢常见,B超、MRI提示40%存在肾上腺增生
◇ DHEA-S通常正常,但TA和雄烯二酮升高
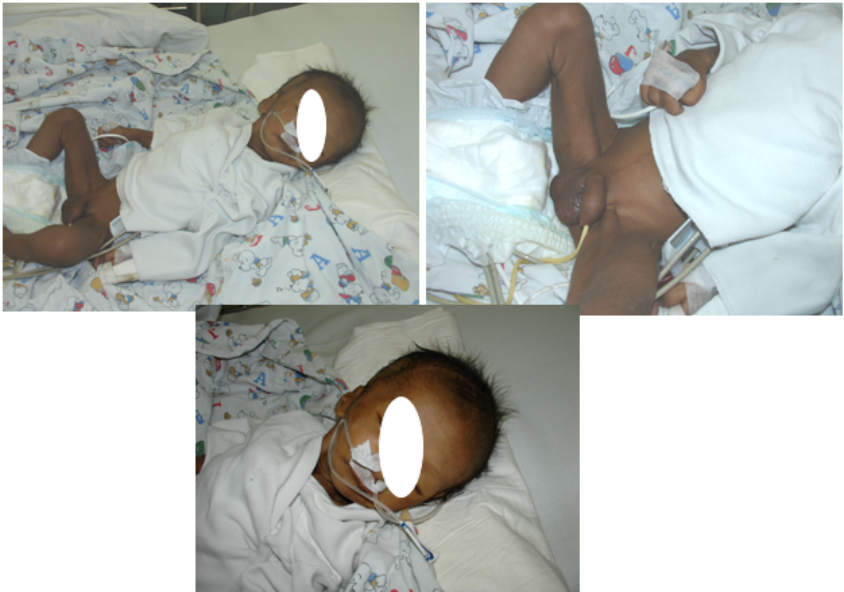
病例
◇ 男,31天
◇ 生后体重不增,纳差入院
◇ PH 7.263 Na 105mmol/l K 5.5mmol/l
◇ Cl 75mmol/l BE –6.7mmo/l
◇ F 5.89μg/dl ACTH >1250pg/ml
◇ T 74.8ng/dl
◇ 24小时尿17-KS 1.26mg 17α-OHP>20ng/ml
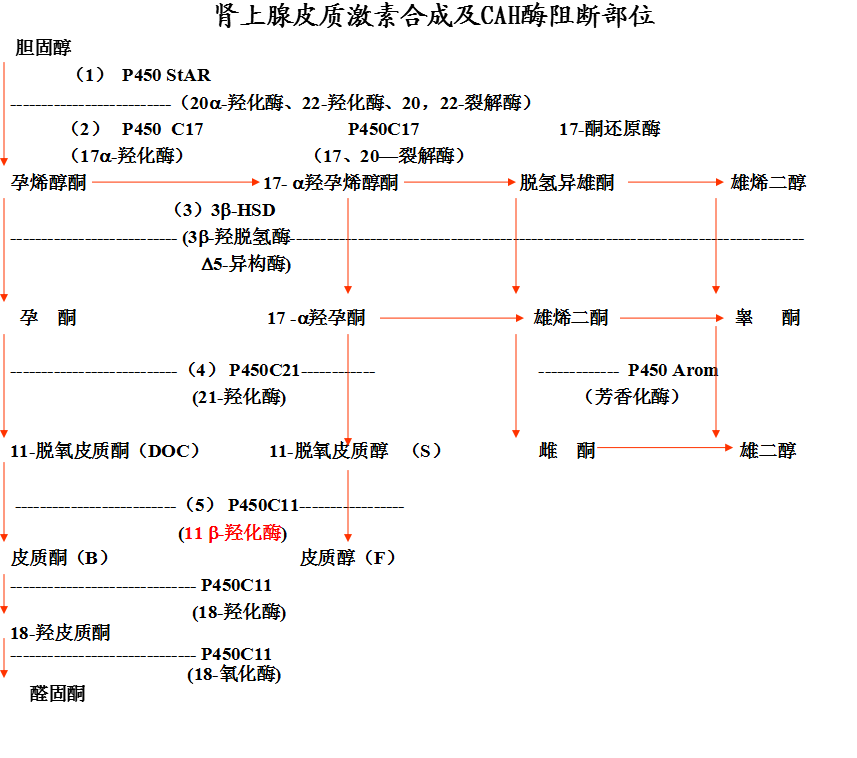
11-β羟化酶缺陷
◇ 占 5-10%
◇ 基因 8q.
◇ 临床特点是低血肾素水平,血11-脱氧皮质醇和11-脱氧皮质酮增高。
◇ 由于脱氧皮质酮的盐皮质激素活性,患者表现为水钠储留,高血压和低钾性碱中毒
◇ 血雄激素水平增加可以导致胎儿雄性化
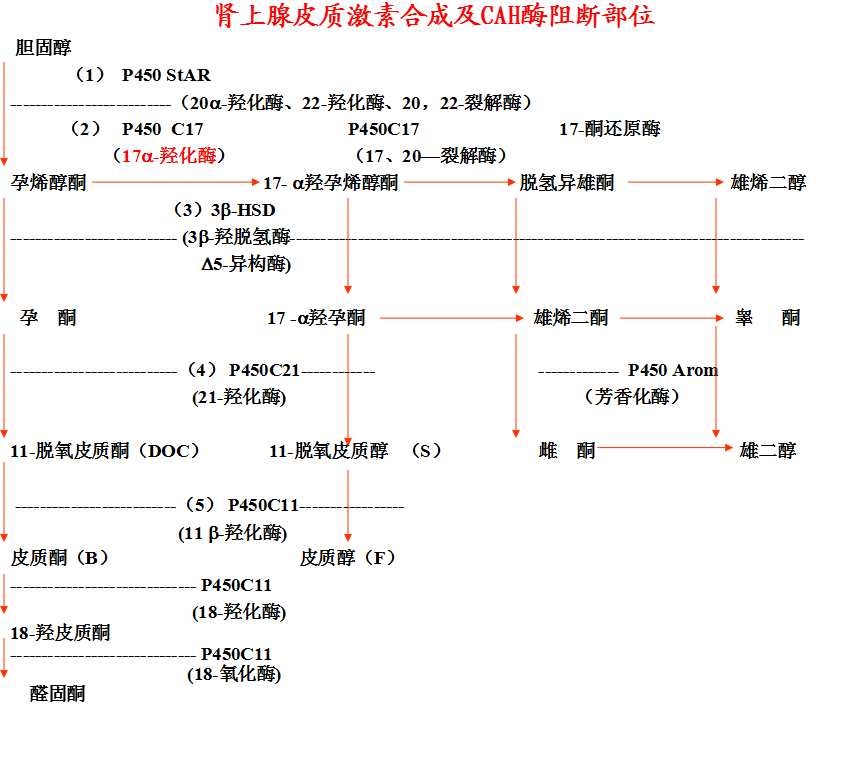
17-a-羟化酶缺陷
◇ 基因位于10号染色体.
◇ 临床表现与 11-羟化酶缺陷类似,但雄激素水平不高,女性没有雄性化表现,男孩二性畸形
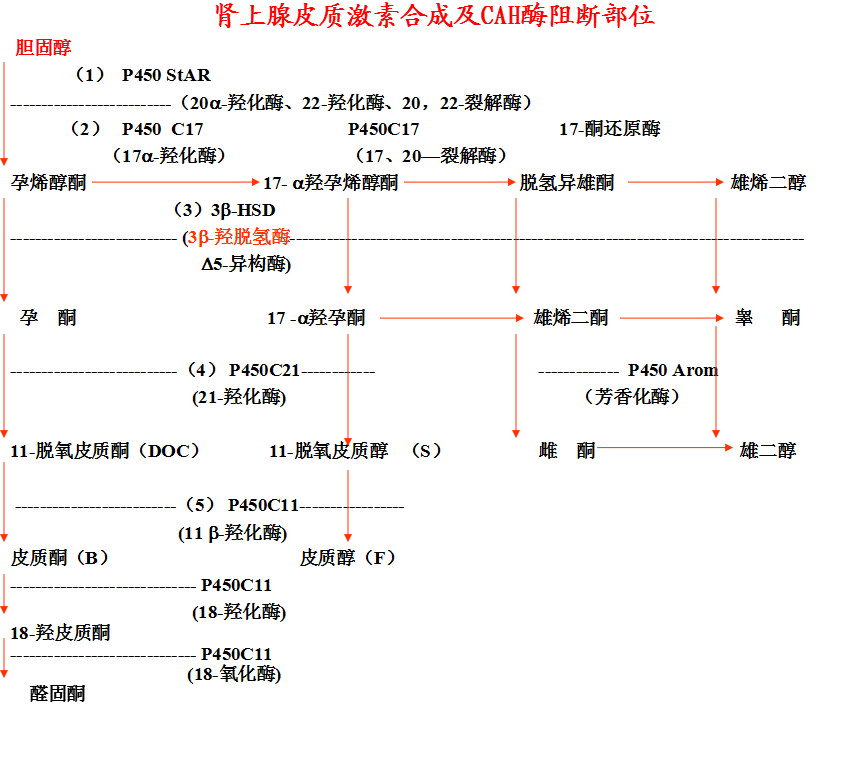
3-羟类固醇脱氢酶缺乏
◇ 发病罕见,17-羟孕烯醇酮、DHEA增加, 在外周组织中转化为睾酮
◇ 典型:失盐和肾上腺功能均不足
◇ 生后2周左右出现恶心、呕吐、脱水、低钠、酸中毒,常因酸中毒死亡
◇ 女婴子宫雄性化和二性畸形,男性不同程度外生殖器发育不良,重者假二性畸形、外生殖器女性化

